«Благодарна дочери за то, что она родилась живой и успела побыть с нами»
В реанимации у Ксюши родители бывали постоянно, выходили только, когда врачи просили их об этом. Девочке постепенно становилось хуже. Еще во время беременности, наблюдая за малышкой на УЗИ, врачи с каждым месяцем отмечали ухудшение ее состояния, пороки сердца, кишечника становились все заметнее. Были и другие признаки синдрома Эдвардса, например, сжатые в кулачки ручки с отставленным в сторону указательным пальчиком.
На третий день Ксюша ушла из жизни. «Нас в этот момент не было рядом. В палату зашел доктор, сказал: «К сожалению, ваш ребеночек умер. Подготовьтесь, если вы хотите попрощаться, возьмите все, что нужно и приходите, мы вам дадим эту возможность».
«Все что нужно» было припасено у Юлии заранее. Она купила красивую одежду, чепчик, взяла с собой икону. Для прощания им с мужем и дочке выделили отдельную комнату, в которой семью никто не тревожил. Для России это новый и пока непривычный опыт, хотя мы знаем, что на Западе такие комнаты при реанимациях существуют уже давно.
«Ксюшу принесли запеленутую, головочка одна торчала, как у матрешки. Она улыбалась нам. У нее было такое милое лицо, потому что уже не было трубок, все время до этого я ее видела с трубочкой. Мы Ксюшу распеленали, одели, понянчили. Гладили, смотрели на нее. Поверьте, это очень важно. Может быть тот, кто с этим не сталкивался, подумает: как можно все это делать с мертвым ребенком? Но это были самые драгоценные минуты», — делится Юлия.
Она добавляет, что очень благодарна дочери за то, что та родилась живой и успела побыть рядом с родителями. «Дом с маяком» подарил мне кулон с именем Ксюшеньки, я ношу его на цепочке рядом с крестиком». На память о девочке также осталась коробочка с дорогими маминому сердцу предметами, среди которых розовое одеяло.
После. «Мы ни на секунду не пожалели, что встретились с нашей дочкой»
Когда все закончилось, все только начинается. Семья остается в числе подопечных «Дома с маяком», с ней работает психолог.
«На этапе беременности часто получается так, что у семьи почти нет потребности в общении с психологом. Они собирают силы и находятся в ожидании. В этот момент для мамы и папы важно, что мы просто рядом, что мы принимаем их выбор, обсуждаем возможности, для нас важна, ценна жизнь их ребенка.
Они собирают силы и находятся в ожидании. В этот момент для мамы и папы важно, что мы просто рядом, что мы принимаем их выбор, обсуждаем возможности, для нас важна, ценна жизнь их ребенка.
Основная работа переживания начинается после смерти малыша, и вот тут специалист подключается сразу – сначала удаленно, по телефону, затем, когда мама окрепнет, в формате очных консультаций», — объясняет Оксана Попова.
Помощь может быть оказана не только родителям, но и всем членам семьи. За консультацией в «Дом с маяком» обращаются и бабушки, и старшие братья и сестры ушедшего ребенка, поскольку в фонде уже налажена работа с сиблингами.
«Мы постоянно учимся у наших подопечных. Скажем, одна мама, когда ждала свою особенную дочку, говорила другим детям – их в семье трое – что девочка родится ангелом, и никто пока не знает, как сложится ее судьба. Возможно, она сразу улетит на небо, а возможно, побудет немного с ними. Это оказалось очень удачной и понятной для детей формулировкой», — комментирует Оксана.
«Перед Новым 2020 годом мы поздравляли все семьи, бывшие в программе, и получили много теплых слов в ответ. Нам было важно и очень приятно получать их фото, на которых мама, папа и старшие дети улыбаются. Видно, что они счастливы, они живут и радуются. И они присылают эту фотографию нам – тем, кто знает, через что они прошли, тем, кто знает их умершего ребенка.
Одна мама написала, что они с мужем ни на секунду не пожалели, что встретились со своей дочкой. Другая призналась, что «этот год был очень хорошим, потому что теперь у нее есть сын», и хотя он уже умер, мама говорит, что он есть».
Интересно, что отношение к перинатальной паллиативной программе незаметно меняется даже в обществе. Если на самом ее старте под постами по этой теме в фейсбуке директора благотворительного фонда «Дом с маяком» Лиды Мониава было много негатива, то сейчас его гораздо меньше, а больше слов поддержки в адрес родителей и тех, кто им помогает.
Важно сказать, что везде, где подобные программы уже существуют – и в США, и в Европе — через них проходит не так много родителей.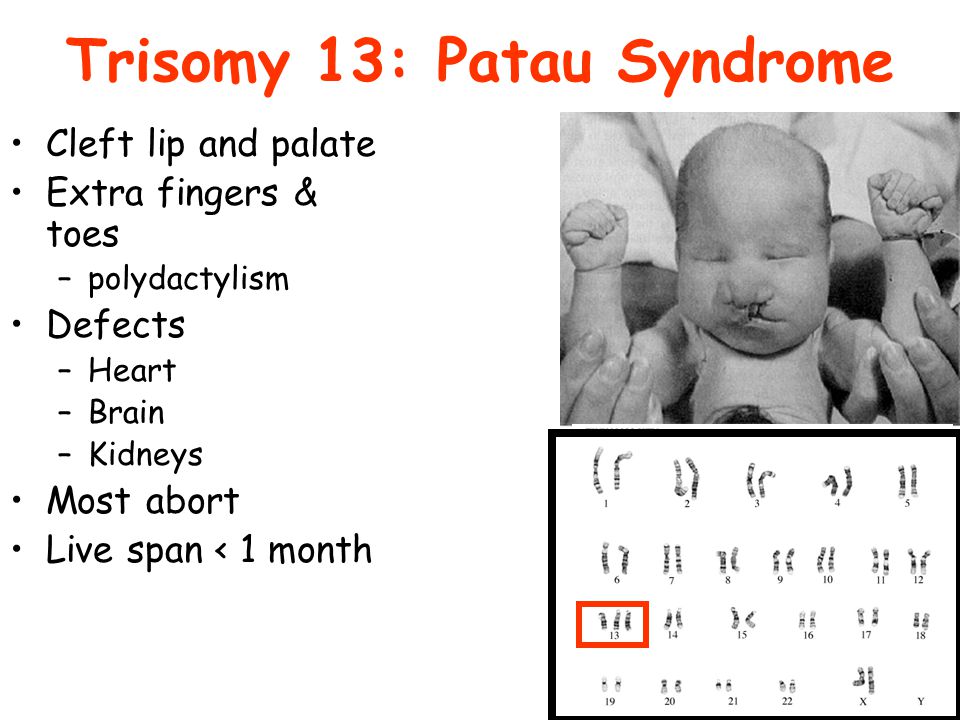 Но то, что теперь и у родителей в Москве и Московской области есть возможность встретиться со своим безнадежно больным ребенком и хотя бы несколько минут, часов или дней побыть рядом с ним, почувствовать себя мамой и папой – большая победа.
Но то, что теперь и у родителей в Москве и Московской области есть возможность встретиться со своим безнадежно больным ребенком и хотя бы несколько минут, часов или дней побыть рядом с ним, почувствовать себя мамой и папой – большая победа.
У перинатальной паллиативной программы много планов. Прежде всего, наладить взаимодействие с роддомами Москвы и Московской области, проводить обучающие мероприятия для врачей, чтобы они знали о возможностях при сохранения проблемной беременности и могли сообщить об этом своим пациентам. Специалисты «Дома с маяком» также планируют изучать зарубежный опыт и находить новые способы поддержки родителей и сохранения памяти об их детях.
Скрининговое исследование плода в I триместре
В 90% случаев у переболевших covid выявляют остаточные явления изменения легких. Наиболее распространен фиброз, когда в легких появляется рубцовая ткань и нарушается насыщение крови кислородом. При ковид-инфекции также могут поражаться почки, печень, желудочно-кишечный тракт, сердце.
Распространенные последствия
Поражение легких:
- одышка при физической нагрузке
- кашель
- повышенная температура тела в пределах 37.0° — 37.5° продолжительное время
опасное для жизни состояние, в результате образования тромбов может развиваться инсульт, инфаркт миокарда или тромбоэмболия.
Заболевания сердца:
нарушения ритма, воспаление миокарда, проявляющимися тяжестью в области сердца, перебоями в работе сердца.
Заболевание почек:
нарушение выделительной функции, развитие почечной недостаточности.
Расстройство нервной системы:
головная боль, нарушение зрения, рассеянность, снижение памяти, снижение концентрации внимания, нарушение сна, чувство страха, депрессия.
Расстройство желудочно-кишечного тракта:
боли в животе, тяжесть в правом подреберье, тошнота, нарушение пищеварения.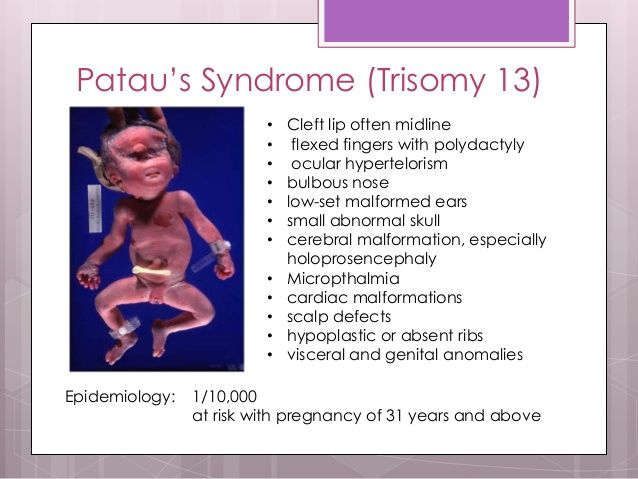
Слабость и боль в мышцах
Мужское бесплодие
Консультация врачом пульмонологом
- Исследование функции внешнего дыхания (ФВД)
- Лабораторную диагностику
- Пульсоксиметрия
Программа реабилитации после Ковид-19
Программа может корректироваться и дополняться лечащим врачом с учетом состояния пациента и особенности перенесенного заболевания.
Индивидуальный подбор лекарственных препаратов
Дыхательная гимнастика
Позволяет улучшить вентиляцию лёгких и насыщения крови кислородом. Регулярное выполнение упражнений увеличивает объём лёгких.
ВЛОК (внутривенное лазерное облучение крови)
Оздоровление организма при помощи действия световой энергии на кровь непосредственно в самих сосудах. Процедура проводится с помощью аппарата — АЛТ «Матрикс-ВЛОК». Сеанс длится 15-20 мин. Помогает улучшить свойства крови, повысить иммунитет, снизить отёчность, снабдить органы кислородом, восстановить обмен веществ.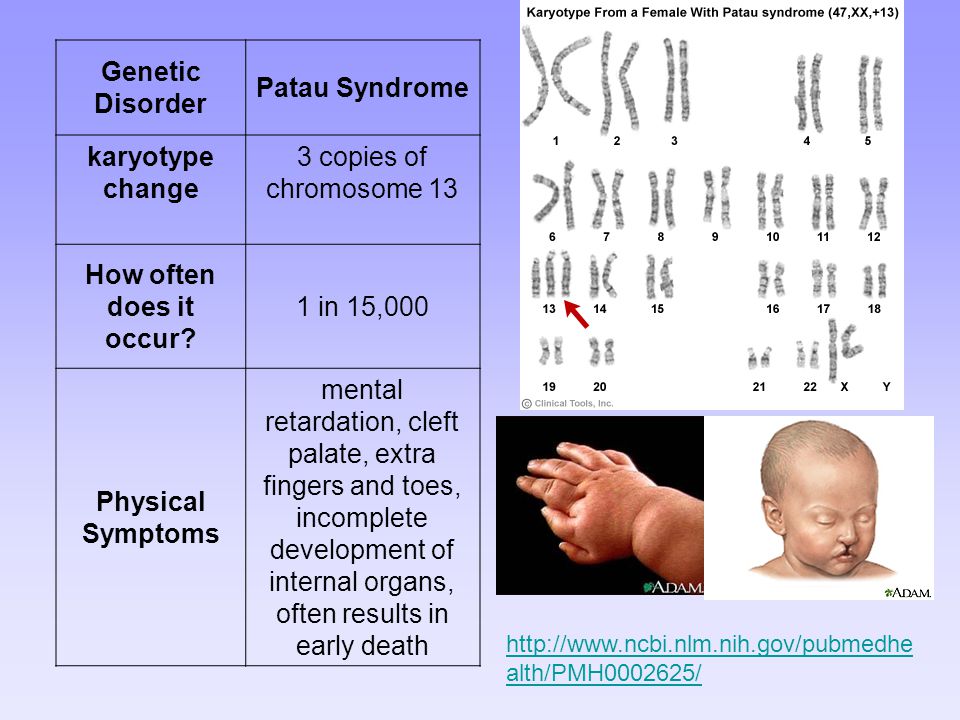
Ингаляции
Лечение, основанное на вдыхании паров необходимых лекарств через небулайзер в дыхательные пути. Местный эффект от использования ингалятора наступает моментально.
Лечебный массаж
Нормализует работу дыхательной системы. Массирующие движения обеспечивают повышение интенсивности кровоснабжения и проходимости бронхов.
Рефлексотерапия
Лечебный способ воздействия на определённые акупунктурные точки организма при помощи специальных игл. Активизирует и восстанавливает внутренние природные силы организма человека.
Комплексная реабилитация позволит полностью восстановиться
- Уменьшить очаги воспаления в кратчайшие сроки;
- Улучшить вентиляцию легких;
- Нормализовать поступления кислорода в организм;
- Устранить обструкцию бронхов;
-
Предотвратить формирование фиброзной ткани, как основной причины развития дыхательной недостаточности после пневмонии.


Реабилитация необходима абсолютно всем пациентам, перенесшим коронавирусную инфекцию, пневмонию и ОРВИ, для восстановления и сохранения качества жизни!
Программу ведут
Страница не найдена — Саянский медицинский колледж
Я, субъект персональных данных, в соответствии с Федеральным законом от 27 июля 2006 года № 152 «О персональных данных» предоставляю ОГБПОУ «Саянский медицинский колледж» (далее — Оператор), расположенному по адресу Иркутская обл., г.Саянск, м/он Южный, 120, согласие на обработку персональных данных, указанных мной в форме веб-чата, обратной связи на сайте в сети «Интернет», владельцем которого является Оператор.
- Состав предоставляемых мной персональных данных является следующим: Имя, адрес электронной почты.
- Целями обработки моих персональных данных являются: обеспечение обмена короткими текстовыми сообщениями в режиме онлайн-диалога или обмена текстовыми сообщениями через электронную почту.
- Согласие предоставляется на совершение следующих действий (операций) с указанными в настоящем согласии персональными данными: сбор, систематизацию, накопление, хранение, уточнение (обновление, изменение), использование, передачу (предоставление, доступ), блокирование, удаление, уничтожение, осуществляемых как с использованием средств автоматизации (автоматизированная обработка), так и без использования таких средств (неавтоматизированная обработка).
- Я понимаю и соглашаюсь с тем, что предоставление Оператору какой-либо информации о себе, не являющейся контактной и не относящейся к целям настоящего согласия, а равно предоставление информации, относящейся к государственной, банковской и/или коммерческой тайне, информации о расовой и/или национальной принадлежности, политических взглядах, религиозных или философских убеждениях, состоянии здоровья, интимной жизни запрещено.
- В случае принятия мной решения о предоставлении Оператору какой-либо информации (каких-либо данных), я обязуюсь предоставлять исключительно достоверную и актуальную информацию и не вправе вводить Оператора в заблуждение в отношении своей личности, сообщать ложную или недостоверную информацию о себе.
- Я понимаю и соглашаюсь с тем, что Оператор не проверяет достоверность персональных данных, предоставляемых мной, и не имеет возможности оценивать мою дееспособность и исходит из того, что я предоставляю достоверные персональные данные и поддерживаю такие данные в актуальном состоянии.
- Согласие действует по достижении целей обработки или в случае утраты необходимости в достижении этих целей, если иное не предусмотрено федеральным законом.
- Согласие может быть отозвано мною в любое время на основании моего письменного заявления.

FISH-диагностика (хромосомы X и Y)
Исследование половых хромосом высокочувствительным молекулярно-цитогенетическим FISH(fluorescence in situ hybridization)-методом позволяет выявить даже небольшие изменения как в хромосоме, так и в ее части (подтвердить наличие хромосомной перестройки, уточнить точки разрыва хромосом и др.).
Синонимы русские
FISH-тест на генетические аномалии половых хромосом, FISH-диагностика плода, FISH-диагностика синдрома Клайнфелтера, FISH-диагностика синдрома Тернера, FISH-диагностика синдрома Мартина — Белл (синдром ломкой X-хромосомы), предимплантационная генетическая диагностика (ПГД), FISH-диагностика заболеваний, сцепленных с полом.
Синонимы английские
FISH analysis on Fragile X Syndrome, FISH diagnosis of 47, XYY Syndrome, FISH-test for genetic abnormalities, FISH analysis of sex chromosomes (X and Y), FISH testing Turner’s Syndrome, FISH diagnosis of Klinefelter’s Syndrome, fetal sex test, preimplantation genetic diagnosis (PGD), FISH diagnosis of Sex-Linked Genetic Diseases.
Метод исследования
Дифференциальное окрашивание хромосом.
Какой биоматериал можно использовать для исследования?Венозную кровь.
Как правильно подготовиться к исследованию?
- Исследование проводится в состоянии сытости, не рекомендуется сдавать кровь на данное исследование натощак.
- Исключить (по согласованию с врачом) прием антибактериальных и химиотерапевтических препаратов в течение 14 дней до исследования.
- Исследование рекомендуется проводить не ранее чем через 2 недели после перенесенных инфекционных/острых воспалительных заболеваний.
Общая информация об исследовании
Цитогенетический анализ проводится методом флуоресцентной гибридизации in situ (FISH, от англ. fluorescence in-situ hybridization). Подробнее с методом можно ознакомиться здесь (https://helix.ru/kb/item/12-052).
У человека 46 хромосом (23 пары), из них две половые — XX или XY. В норме у женщины имеется 2 X хромосомы, такой кариотип обозначается как 46XX, у мужчины есть одна X и одна Y хромосома (кариотип 46XY). Возможны различные варианты количественных и качественных генетических аномалий половых хромосом. Например, приблизительно у 1 из 350 новорождённых мальчиков кариотип 47,XXY или 47,XYY, а у одного ребенка на каждые несколько тысяч новорождённых — моносомия по Х-хромосоме.
Аномалии половых хромосом являются общими и вызывают синдромы, связанные с рядом физических и психических нарушений. Многие из этих заболеваний не определяются внутриутробно, если беременной не проводится пренатальное тестирование по другим причинам, например из-за ее более старшего возраста.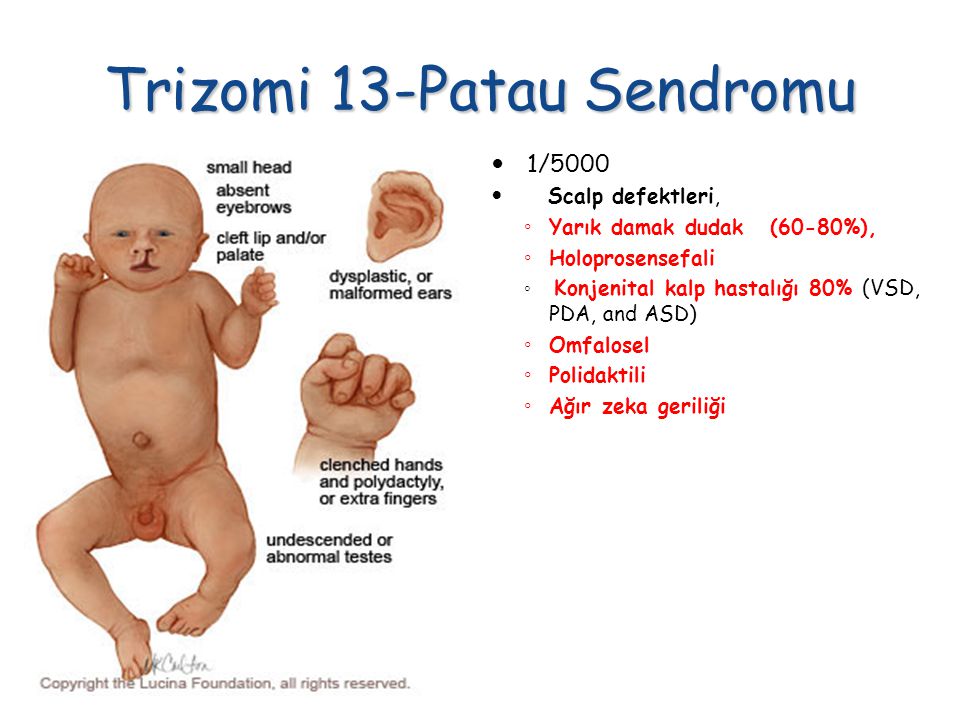 Часто отклонения трудно распознать при рождении и они диагностируются только в период пубертата.
Часто отклонения трудно распознать при рождении и они диагностируются только в период пубертата.
Синдромы, вызванные аномалиями половой хромосомы, менее выражены, чем при патологии аутосомных хромосом. Например, девочки, у которых есть дополнительная Х, часто кажутся нормальными физически и умственно, затем, вырастая, являются плодовитыми. Напротив, у детей с лишними аутосомными хромосомами (от 1 до 22) обычно наблюдаются серьезные нарушения, такие как синдром Дауна, который обычно возникает при трисомии хромосомы 21 (три вместо пары). А дополнительная хромосома 1 может быть фатальной для плода. Девочки без половой хромосомы жизнеспособны, тогда как плоды, у которых отсутствует аутосомная хромосома, не выживают. Часто сниженная фертильность или бесплодие связаны с патологией в половой хромосоме. Так, при первичной аменорее аберрации Х-хромосомы находят примерно у 25% женщин. В связи с этим многим бесплодным парам рекомендуется пройти генетическое исследование.
В настоящее время существует более 300 наследственных заболеваний, передающихся сцепленно с полом (например, Болезнь Фабри, гемофилия А и В, прогрессирующие мышечные дистрофии Дюшенна и Беккера, X-сцепленная глухота).
Синдром Тернера (в РФ более известный как синдром Шерешевского — Тернера) встречается примерно у 1/2500-3000 живых женских родов во всем мире. 99% из 45,X0 вариантов прерываются спонтанно. Около 50% пораженных девочек имеют 45,X0 кариотип, около 80% из них потеряли отцовскую X. Большинство других из 50% — это мозаики (например, 45,X0/46,XX или 45,X0/47,XXX). Около 15-20% из половины случаев связаны со структурными перестройками Х-хромосомы, например делецией короткого или длинного плеча, изохромосомой Х по длинному или короткому плечу, кольцевой Х-хромосомой. Среди мозаичных девочек фенотип может отличаться от типичного для этой патологии.
Среди мозаичных девочек фенотип может отличаться от типичного для этой патологии.
Беременность плодом, имеющим синдром Тернера, часто протекает неблагоприятно, с угрозой выкидыша и преждевременных родов. И как правило, риск возникновения этой генетической поломки никак не связан с возрастом беременной. У младенцев высокий риск развития дисплазии бедра, а 10% подростков имеют сколиоз. Остеопороз и переломы довольно часто отмечаются среди женщин с этим синдромом. Часто новорождённые с синдромом Тернера практически не отличаются от здоровых детей. но у многих из них даже при доношенном сроке беременности наблюдается сниженная масса тела и небольшой рост. Некоторые из них имеют выраженную отечность рук и ног, лимфостаз и/или короткую шею со свободными складками кожи по бокам (птеригиум-синдром). В раннем возрасте часто наблюдается слабый сосательный рефлекс, моторное беспокойство, срыгивания фонтаном, отставание в физическом развитии. Умственная отсталость встречается редко, но у многих детей есть проблемы с обучением из-за дефицита внимания и/или гиперактивности. При классическом типе заболевание проявляется коарктацией аорты и различными врождёнными пороками сердца. Артериальная гипертензия часто возникает в старшем возрасте, даже без коарктации. Также характерны аномалии почек (например, подковообразная) и гемангиомы. У большинства пациентов наблюдаются потеря слуха, косоглазие, дальнозоркость или близорукость, дальтонизм. Дисгенезия гонад (вместо яичников лентовидные полоски белесоватой ткани без ооцитов) характерна для 90% женщин. Тиреоидит, гипотиреоз, сахарный диабет, алопеция, ожирение, гипертрихоз и целиакия более распространены, чем среди населения в целом. Взрослые обычно низкорослые, с короткой шеей с крыловидными складками, широкой грудной клеткой, низкой границей роста волос, с недоразвитой нижней челюстью, высоким нёбом, аномалиями прикуса, деформацией ушных раковин. Также обращают на себя внимание множественные пигментированные невусы, девиация локтевых суставов, укорочение IV и V пальцев на руках и ногах и гипоплазия ногтей. Дисгенезия гонад приводит к невозможности нормального полового созревания и к соответствующим клиническим симптомам (например, отсутствие менструации, недоразвитие первичных половых признаков).
При классическом типе заболевание проявляется коарктацией аорты и различными врождёнными пороками сердца. Артериальная гипертензия часто возникает в старшем возрасте, даже без коарктации. Также характерны аномалии почек (например, подковообразная) и гемангиомы. У большинства пациентов наблюдаются потеря слуха, косоглазие, дальнозоркость или близорукость, дальтонизм. Дисгенезия гонад (вместо яичников лентовидные полоски белесоватой ткани без ооцитов) характерна для 90% женщин. Тиреоидит, гипотиреоз, сахарный диабет, алопеция, ожирение, гипертрихоз и целиакия более распространены, чем среди населения в целом. Взрослые обычно низкорослые, с короткой шеей с крыловидными складками, широкой грудной клеткой, низкой границей роста волос, с недоразвитой нижней челюстью, высоким нёбом, аномалиями прикуса, деформацией ушных раковин. Также обращают на себя внимание множественные пигментированные невусы, девиация локтевых суставов, укорочение IV и V пальцев на руках и ногах и гипоплазия ногтей. Дисгенезия гонад приводит к невозможности нормального полового созревания и к соответствующим клиническим симптомам (например, отсутствие менструации, недоразвитие первичных половых признаков). В подавляющем большинстве случаев женщины бесплодны, но при мозаичных вариантах возможно зачатие и вынашивание плода.
В подавляющем большинстве случаев женщины бесплодны, но при мозаичных вариантах возможно зачатие и вынашивание плода.
Крайне редко синдром Тернера встречается у мужчин (известно чуть более 70 случаев) и связан в таком случае с транслокацией или хромосомным мозаицизмом.
Синдром Мартина — Белл (синдром ломкой X-хромосомы, fragile X syndrome) является наиболее часто диагностируемой наследственной причиной умеренной умственной недостаточности. При этом чаще страдают мальчики, чем девочки. Симптомы синдрома Мартина — Белл вызваны аномалией гена FMR1 в локусе Хq27.3, приводящей к недостаточной выработке белка FMR1, необходимого для нормального развития нервной системы. Эта патология встречается приблизительно у одного из 2000-3000 мужчин и у одной из 259 женщин. Дети и взрослые могут иметь физические, интеллектуальные и поведенческие проблемы. Новорождённые крупные с большой головой, с широким и высоким лбом, с большими (часто оттопыренными) ушами, вытянутым лицом и выступающим подбородком. Многие из них светловолосые с голубыми глазами. У мальчиков большие яички, что становится наиболее очевидным после полового созревания. Часто наблюдаются аномально гибкие подвижные суставы, возможно развитие сердечной недостаточности из-за пролапса митрального клапана. Могут быть не все признаки, а один или несколько. У всех детей наблюдается олигофрения разной степени выраженности, сопровождающаяся различной неврологической симптоматикой. Могут развиться проявления, напоминающие аутизм (например, непереносимость прикосновений, плохой зрительный контакт, эхолалия). Такие больные часто говорят быстро, сбивчиво, может быть «бормочущая речь», разнообразные гримасы, монотонное хныканье и дискоординация движений.
Многие из них светловолосые с голубыми глазами. У мальчиков большие яички, что становится наиболее очевидным после полового созревания. Часто наблюдаются аномально гибкие подвижные суставы, возможно развитие сердечной недостаточности из-за пролапса митрального клапана. Могут быть не все признаки, а один или несколько. У всех детей наблюдается олигофрения разной степени выраженности, сопровождающаяся различной неврологической симптоматикой. Могут развиться проявления, напоминающие аутизм (например, непереносимость прикосновений, плохой зрительный контакт, эхолалия). Такие больные часто говорят быстро, сбивчиво, может быть «бормочущая речь», разнообразные гримасы, монотонное хныканье и дискоординация движений.
Синдром FXTAS (тремор/атаксия, ассоциированные с ломкой Х-хромосомой) может поражать до 1 из 3000 мужчин старше 50 лет. Он является результатом менее обширной аномалии (называемой премутацией) в гене FMR1. Риск развития расстройства возрастает по мере старения.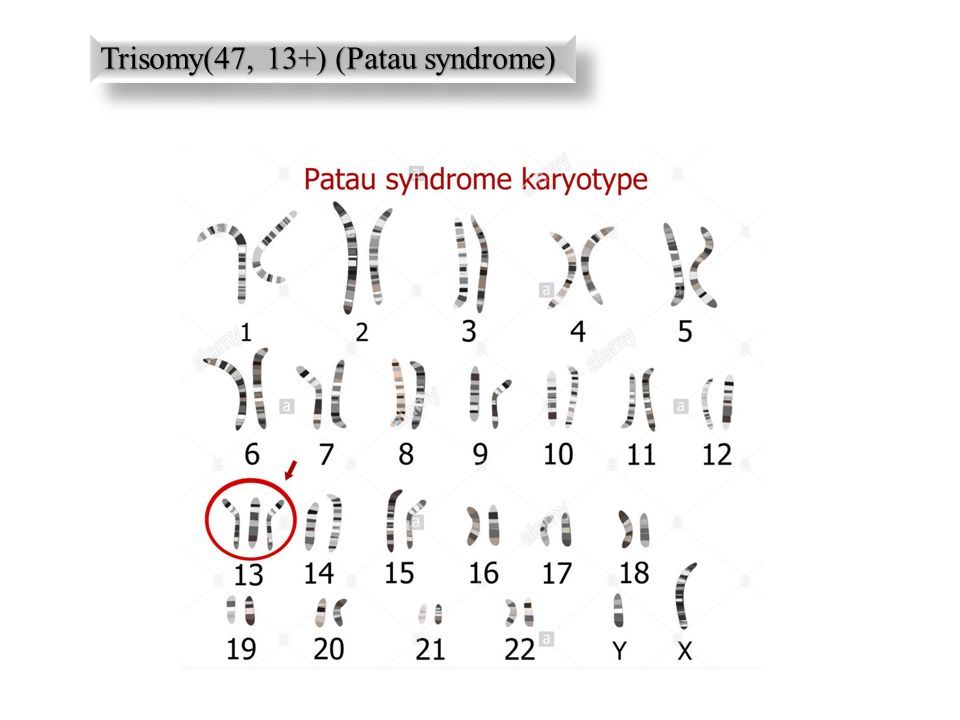 Часто заболевание начинается с тремора рук во время выполнения какого-то движения. Затем появляются проблемы с координацией (медленно прогрессирует атаксия), паркинсонизм и в конечном итоге деменция. На поздних стадиях может утрачиваться контроль над функциями тазовых органов. После появления симптомов люди могут прожить от пяти до двадцати пяти лет.
Часто заболевание начинается с тремора рук во время выполнения какого-то движения. Затем появляются проблемы с координацией (медленно прогрессирует атаксия), паркинсонизм и в конечном итоге деменция. На поздних стадиях может утрачиваться контроль над функциями тазовых органов. После появления симптомов люди могут прожить от пяти до двадцати пяти лет.
При синдроме тройного Х (Triple X) дополнительная Х-хромосома обычно унаследована от матери. Чем старше мать, тем больше риск развития у плода этого синдрома. Примерно 1 из каждых 1000 девочек рождается с третьей Х-хромосомой. Синдром Triple X редко вызывает очевидные физические нарушения. Девочки могут иметь более низкий уровень интеллекта, проблемы с вербальными навыками и больше проблем с обучением, чем их братья и сестры. Иногда синдром вызывает нарушения менструального цикла и бесплодие. Тем не менее некоторые женщины с синдромом тройного Х родили физически нормальных детей с нормальным кариотипом. По данным некоторых исследований, около 90% трисомиков по X-хромосоме остаются невыявленными.
В чрезвычайно редких случаях рождаются младенцы с четырьмя или даже пятью Х-хромосомами. Чем больше Х-хромосом, тем больше вероятность умственной отсталости и физических аномалий.
Синдром 47, XYY встречается примерно у 1/1000 мальчиков. Дети, как правило, выше среднего и имеют небольшое снижение IQ по сравнению с членами семьи. Наличие второй Y-хромосомы в большинстве случаев не ведёт к каким-либо физическим отклонениям. В младшем возрасте могут быть расстройства поведения, гиперактивность, нарушение внимания и расстройства обучения. Взрослые мужчины часто импульсивны, эмоционально незрелы, могут казаться неуклюжими.
Синдром Клайнфелтера — наиболее распространенное расстройство половой хромосомы, встречающееся примерно у 1 из 500 новорождённых мальчиков. Чаще всего он возникает из-за наличия дополнительной копии Х-хромосомы в каждой клетке (47, XXY). В 60% случаев дополнительная Х-хромосома — материнская. Часто это препятствует нормальному функционированию яичек и приводит к снижению уровня андрогенов. Мужчины с этим синдромом, как правило, высокие с непропорционально длинными руками и ногами. У 70% из них развивается гинекомастия (увеличение грудных желез). Половое созревание обычно происходит в срок или с небольшой задержкой, но часто плохо растут усы и борода. Дети с синдромом Клайнфелтера часто имеют трудности с обучением и задержку развития речи. Они могут быть как спокойны, чувствительны и ненавязчивы, так и, наоборот, агрессивными, склонными к асоциальному поведению. По сравнению со здоровыми мужчинами у взрослых с этим синдромом имеется повышенный риск развития рака молочной железы, системной красной волчанкой и легочных заболеваний. Развитие яичек варьируется от полностью нефункциональных канальцев до некоторого производства сперматозоидов; часто повышается экскреция фолликулостимулирующего гормона с мочой. Примерно в 15% случаев наблюдается мозаицизм, который сопровождается менее выраженной клиникой, дает лучший прогноз в отношении фертильности и психосоциальной адаптации. Встречаются мужчины с синдромом Клайнфелтера, у которых есть 3, 4 и даже 5 Х хромосом.
Мужчины с этим синдромом, как правило, высокие с непропорционально длинными руками и ногами. У 70% из них развивается гинекомастия (увеличение грудных желез). Половое созревание обычно происходит в срок или с небольшой задержкой, но часто плохо растут усы и борода. Дети с синдромом Клайнфелтера часто имеют трудности с обучением и задержку развития речи. Они могут быть как спокойны, чувствительны и ненавязчивы, так и, наоборот, агрессивными, склонными к асоциальному поведению. По сравнению со здоровыми мужчинами у взрослых с этим синдромом имеется повышенный риск развития рака молочной железы, системной красной волчанкой и легочных заболеваний. Развитие яичек варьируется от полностью нефункциональных канальцев до некоторого производства сперматозоидов; часто повышается экскреция фолликулостимулирующего гормона с мочой. Примерно в 15% случаев наблюдается мозаицизм, который сопровождается менее выраженной клиникой, дает лучший прогноз в отношении фертильности и психосоциальной адаптации. Встречаются мужчины с синдромом Клайнфелтера, у которых есть 3, 4 и даже 5 Х хромосом.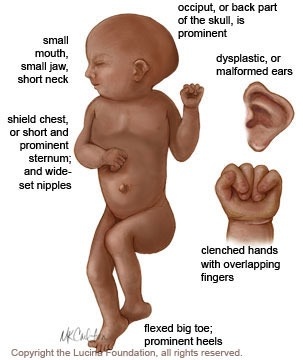 По мере увеличения количества хромосом X возрастает также выраженность умственной отсталости и пороков развития. Каждый дополнительный X связан с сокращением IQ на 15-16 единиц, с речевыми нарушениями. Диагноз «синдром Клайнфелтера» подозревается при физическом осмотре подростка с маленькими яичками и гинекомастией. У многих мужчин он диагностируется во время оценки бесплодия (вероятно, все немозаичные 47, XXY мужчины бесплодны).
По мере увеличения количества хромосом X возрастает также выраженность умственной отсталости и пороков развития. Каждый дополнительный X связан с сокращением IQ на 15-16 единиц, с речевыми нарушениями. Диагноз «синдром Клайнфелтера» подозревается при физическом осмотре подростка с маленькими яичками и гинекомастией. У многих мужчин он диагностируется во время оценки бесплодия (вероятно, все немозаичные 47, XXY мужчины бесплодны).
Для чего используется исследование?
- Для диагностики генетических нарушений половых хромосом.
Когда назначается исследование?
- При бесплодии.
- При привычном невынашивании плода.
- При измененном кариотипе абортивного материала.
- При множественных неудачных попытках ЭКО.
- При предимплантационной генетической диагностике (ПГД) в рамках ЭКО.
- Если во время классического кариотипирования возникли подозрения, требующие уточнения.
- Пренатальная диагностика при подозрении на наличие отклонений в развитии плода (например, отклонения от нормы во время УЗИ).
- При возможном влиянии мутагенных факторов во время беременности.
- Постнатальная диагностика генетической патологии у ребенка при наличии соответствующих клинических признаков.
- При планировании последующих беременностей, если в семье есть ребенок с хромосомной аномалией.

Что означают результаты?
В норме двадцать третья пара хромосом (половая) — это XY у мужчины и ХХ у женщины.
При исследовании эякулята в каждом сперматозоиде должен быть один сигнал половой хромосомы (либо X, либо Y).
В настоящее время 56% морфологически нормальных эмбрионов женщин после 35 имеют хромосомные аномалии.
Что может влиять на результат?
Во время пренатальной диагностики есть риск «засорения» образца материнскими клетками, что может повлиять на результат исследования.
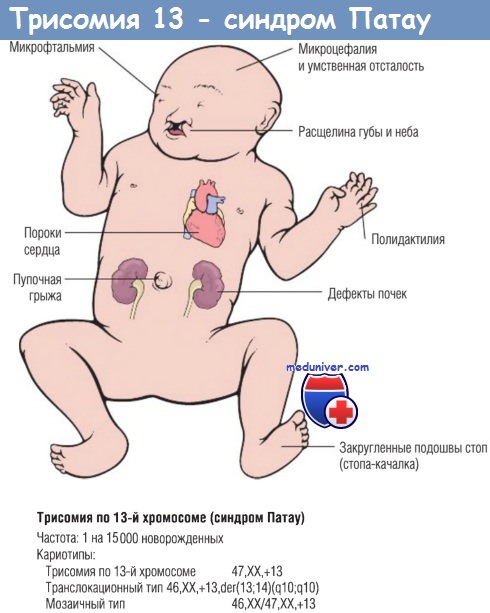
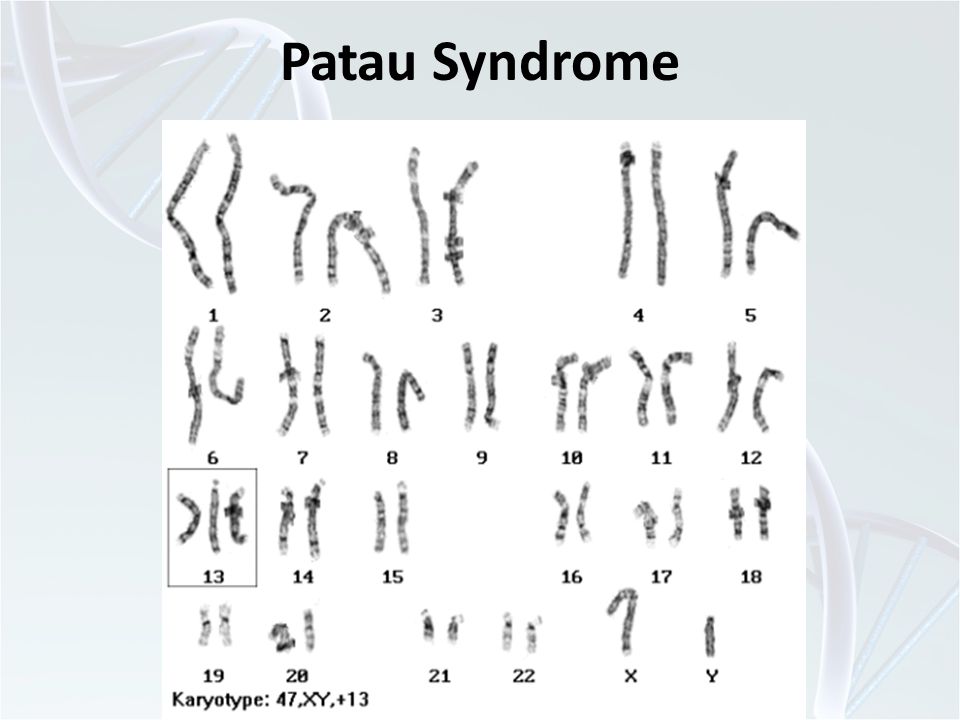
Синдром Патау (трисомия 13) — хромосомное заболевание человека, которое характеризуется наличием в клетках дополнительной хромосомы 13.
Синдром Патау (трисомия 13) — хромосомное заболевание человека, которое характеризуется наличием в клетках дополнительной хромосомы 13. Синдром Патау – генетическая аномалия, для которой характерно наличие у ребёнка многочисленных пороков строения и функционирования органов и систем, головного мозга и опорно-двигательного аппарата. Обуславливается возникновение такого расстройства наличием запасной тринадцатой хромосомы. Для недуга характерны дефекты центральной нервной и сердечно-сосудистой систем, органов зрения и слуха, мышечной системы (наличие расщелины губы), патологии строения органов половой системы. Установлено, что причиной синдрома Патау, в большинстве случаев, является утроение 13 хромосомы, то есть в каждой клетке имеется не две (норма), а три копии тринадцатой хромосомы. В очень редких случаях только часть клеток организма имеет дополнительную копию.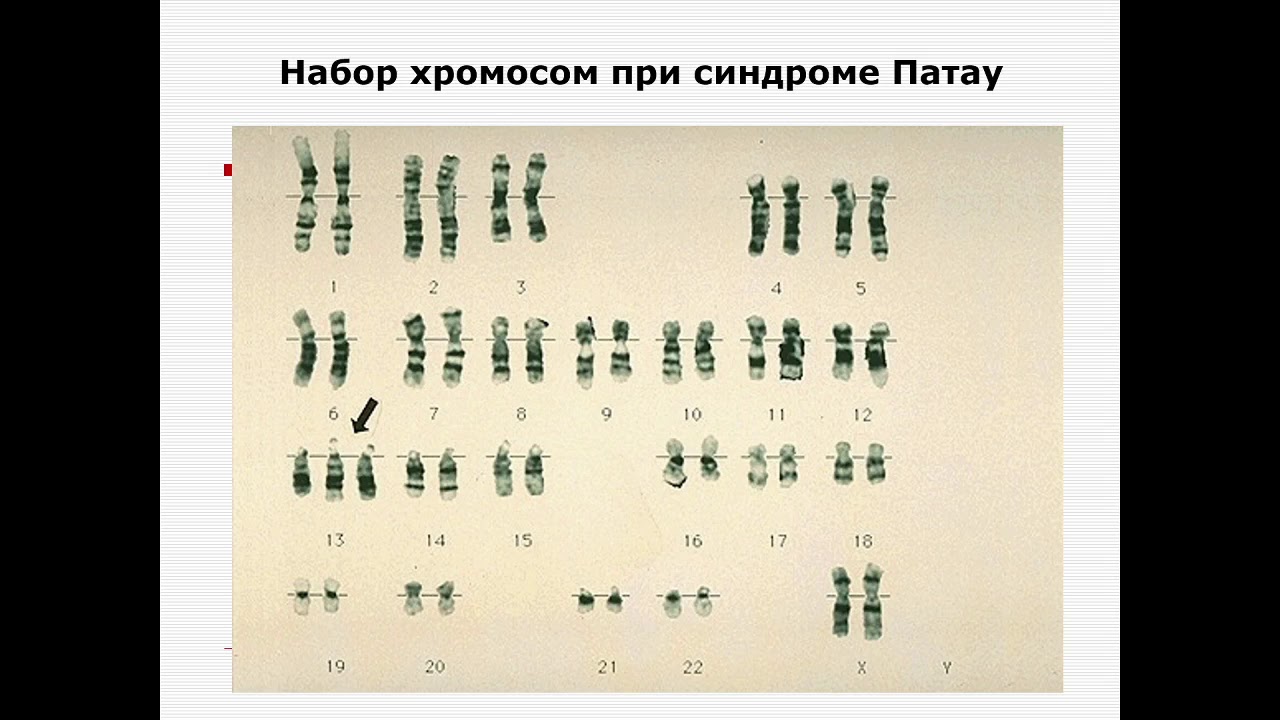 Это так называемый мозаичный синдром Патау. Еще одна причина заболевания – транслокация (перестройка) хромосом, когда часть 13 хромосомы до или непосредственно в момент зачатия привязывается к другой негомологичной хромосоме. В результате такой перестройки у больных наряду с двумя копиями 13 хромосомы появляется дополнительный материал из нее, который подключен к другой хромосоме. Возникает частичная трисомия 13 хромосомы, при которой физические признаки синдрома несколько отличаются от типичной клинической картины. Как правило, синдром Патау не наследуется, а возникает случайно в процессе формирования сперматозоидов и яйцеклеток. Если при делении клеток возникает ошибка, которая называется нерасхождением, это приводит к появлению половых клеток с неправильным числом хромосом. Когда подобные атипичные сперматозоиды и яйцеклетки вовлекаются в генетическую структуру ребенка, он получает дополнительную 13 хромосому во всех клетках организма. Мозаицизм данного синдрома также не наследуется и возникает как случайный сбой при делении клеток на первоначальном этапе развития зародыша.
Это так называемый мозаичный синдром Патау. Еще одна причина заболевания – транслокация (перестройка) хромосом, когда часть 13 хромосомы до или непосредственно в момент зачатия привязывается к другой негомологичной хромосоме. В результате такой перестройки у больных наряду с двумя копиями 13 хромосомы появляется дополнительный материал из нее, который подключен к другой хромосоме. Возникает частичная трисомия 13 хромосомы, при которой физические признаки синдрома несколько отличаются от типичной клинической картины. Как правило, синдром Патау не наследуется, а возникает случайно в процессе формирования сперматозоидов и яйцеклеток. Если при делении клеток возникает ошибка, которая называется нерасхождением, это приводит к появлению половых клеток с неправильным числом хромосом. Когда подобные атипичные сперматозоиды и яйцеклетки вовлекаются в генетическую структуру ребенка, он получает дополнительную 13 хромосому во всех клетках организма. Мозаицизм данного синдрома также не наследуется и возникает как случайный сбой при делении клеток на первоначальном этапе развития зародыша. Далее опишем симптомы болезни Патау, которые можно заметить как во время беременности, так и после рождения малыша. При беременности Синдром Патау имеет следующие признаки во время беременности: •Время пребывания плода в утробе сокращается до 38 недель. •Возникает многоводие (количество околоплодных вод превышает норму). •В процессе вынашивания может быть выкидыш. Ребёнок рождается мёртвым. У новорожденных Далее перечислим характерные симптомы синдрома: •Ребёнок рождается недоразвитым, что проявляется как умственными, так и физическими отклонениями. •Врождённые пороки (мутации и уродства). •Нарушения в развитии нервной системы. •Деформация рук. •Деформация черепа и уродства лица. •Западание переносицы. •Помутнение роговицы глаз.
Далее опишем симптомы болезни Патау, которые можно заметить как во время беременности, так и после рождения малыша. При беременности Синдром Патау имеет следующие признаки во время беременности: •Время пребывания плода в утробе сокращается до 38 недель. •Возникает многоводие (количество околоплодных вод превышает норму). •В процессе вынашивания может быть выкидыш. Ребёнок рождается мёртвым. У новорожденных Далее перечислим характерные симптомы синдрома: •Ребёнок рождается недоразвитым, что проявляется как умственными, так и физическими отклонениями. •Врождённые пороки (мутации и уродства). •Нарушения в развитии нервной системы. •Деформация рук. •Деформация черепа и уродства лица. •Западание переносицы. •Помутнение роговицы глаз.
Что такое наследственные заболевания и как с ними быть?
Наследственные заболевания передаются от одного или обоих родителей детям. Они вызываются генетическими мутациями, но далеко не все генетические заболевания являются наследственными. Как в этом разобраться, какие виды заболеваний бывают, как их лечить и как диагностировать — рассказываем в нашей статье.
Как в этом разобраться, какие виды заболеваний бывают, как их лечить и как диагностировать — рассказываем в нашей статье.
Содержание
Что такое наследственные заболевания?
Наследственные заболевания — это заболевания, обусловленные генными или хромосомными мутациями. У людей от 20 000 до 25 000 генов. Генетическая мутация возникает, когда изменяется один или несколько генов. Если это генетическое изменение передается детям, то это наследственное генетическое заболевание.
При совпадении у партнеров статусов носительства определенных болезней есть высокий риск рождения ребенка с наследственным заболеванием. Если у вас не проявляются симптомы заболевания, вы по-прежнему можете быть носителем и передать мутации своим детям.
Многие генетически обусловленные заболевания проявляются не сразу после рождения, а спустя некоторое время. От наследственных заболеваний следует отличать врожденные заболевания, вызванные внутриутробными повреждениями, например, инфекцией или внешними воздействиями.
Чем отличаются наследственные заболевания от врожденных нарушений?
Генетические заболевания являются результатом изменения одного или нескольких генов и могут передаваться в поколениях или нет.
Все наследственные заболевания имеют генетическое происхождение, т. е. являются результатом изменения одного или нескольких генов и передаются из поколения в поколение. Симптомы могут не проявляться с самого рождения.
Врожденные нарушения могут быть наследственными или нет, а симптомы могут проявляться с рождения. Но их появление не обязательно связано с генетикой.
Виды наследственных заболеваний
Наследственные заболевания разделяются на хромосомные, генные и митохондриальные.
Хромосомные заболевания
В настоящее время описано около 1000 форм хромосомных заболеваний. Хромосомные заболевания возникают в результате изменения числа или структуры хромосом. Они характеризуются общими признаками: маленькая масса и длина тела при рождении, отставание в умственном и физическом развитии, задержка и аномалии полового развития и прочее.
Хромосомные заболевания наследуются редко. И более чем в 95% случаев риск повторного рождения в семье ребенка с хромосомной патологией не превышает общепопуляционного уровня. Хромосомные заболевания с аномалиями числа хромосом включают: синдром Патау, синдром Эдвардса, синдром трисомии хромосомы 8. А хромосомные заболевания с аномалиями структуры хромосом — синдром Ди Джорджи, синдром Вольфа-Хиршхорна, синдром «кошачьего крика», синдром Альфи, синдром Орбели.
Моногенные заболевания
Моногенные заболевания возникают в результате повреждения ДНК на уровне гена. Количество моногенных заболеваний по некоторым оценкам достигает 5000.
Среди признаков моногенных болезней можно выделить: различные формы умственной отсталости, дефекты органов слуха, зрения, скелетные дисплазии, болезни нервной, эндокринной, иммунной и других систем. К числу наиболее известных моногенных болезней относятся муковисцидоз, гемофилия А и В, болезнь Гоше, миодистрофия Дюшенна/Беккера, спинальная мышечная атрофия, дальтонизм.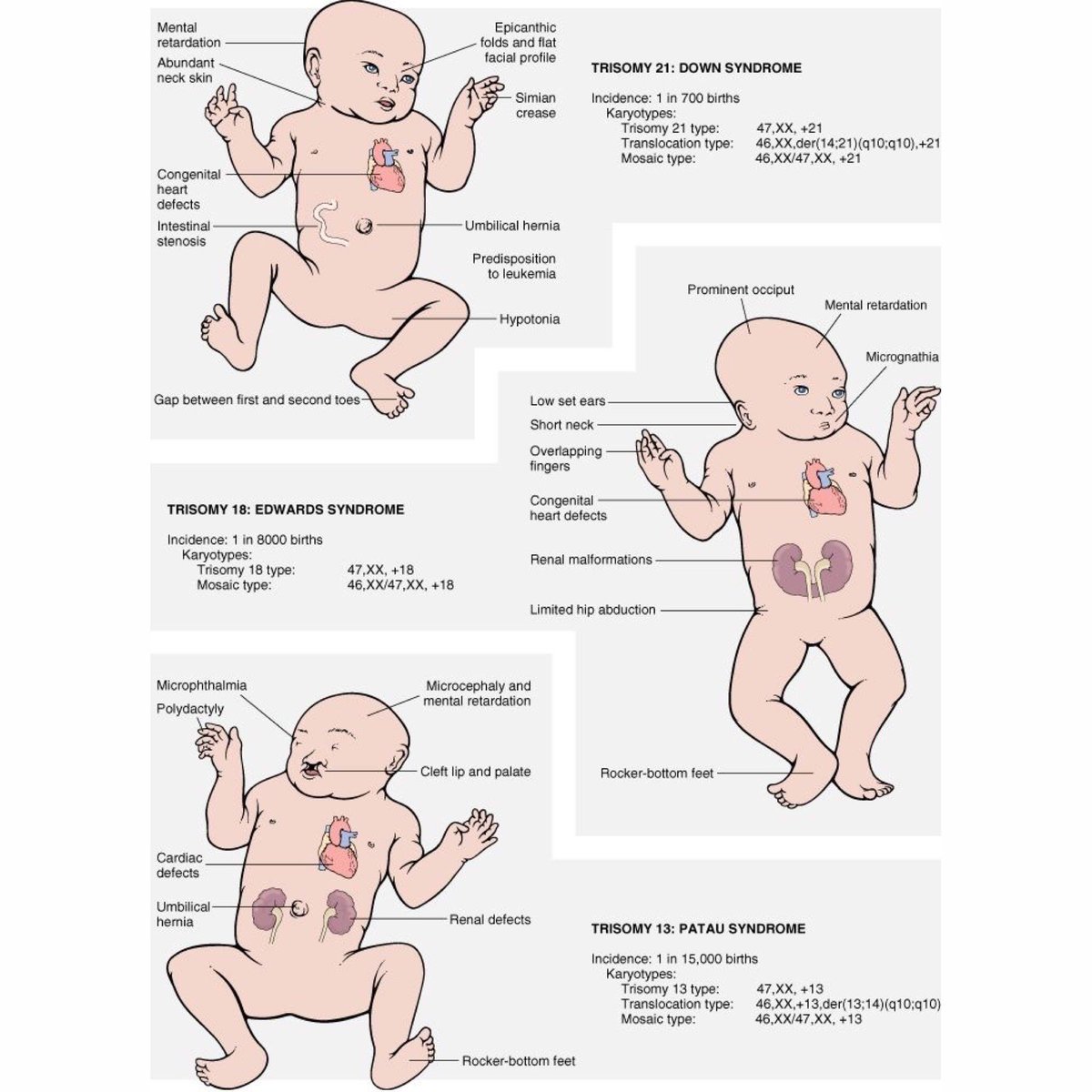
Выявить тяжелые моногенные заболевания можно с помощью пренатальной диагностики, а также, определив наличие мутаций у родителей с помощью генетического теста.
Митохондриальные заболевания
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами в функционировании митохондрий, которые приводят к нарушению тканевого дыхания.
Митохондрии содержат свою собственную ДНК. А болезни, вызванные мутациями в митохондриальной ДНК, наследуются исключительно по материнской линии. Если именно таким образом было унаследовано митохондриальное заболевание, существует 100% вероятность того, что каждый ребенок в семье его унаследует.
Симптомы могут включать в себя: нарушение роста, слабость мышц, аутизм, ментальные расстройства, проблемы с дыханием, слухом и зрением. Примеры митохондриальных заболеваний: синдром Лея, синдром Вольфа-Паркинсона-Уайта, наследственная оптическая нейропатия Лебера и другие.
Полигенные или мультифакториальные заболевания
Существуют также болезни с наследственной предрасположенностью, которые называют мультифакториальными или полигенными заболеваниями.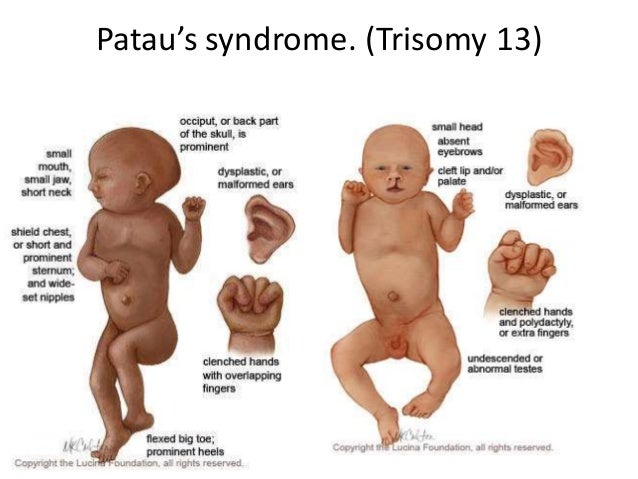
Мультифакториальные заболевания обусловлены наследственными факторами риска, и в значительной степени — неблагоприятным воздействием среды. К мультифакториальным заболеваниям относятся большинство хронических заболеваний, включая сердечно-сосудистые, эндокринные, иммунные, нервно-психические, онкологические и др. Например, бронхиальная астма, сахарный диабет, ревматоидный артрит, гипертоническая болезнь сердца и т.д.
Как передаются наследственные заболевания?
Организм человека состоит из триллионов клеток. Каждая клетка имеет ядро, которое содержит хромосомы. Каждая хромосома состоит из плотно свернутых нитей дезоксирибонуклеиновой кислоты (ДНК).
Гены — это инструкции по сборке белков в нашем организме, которые определяют специфические черты каждого человека, например, цвет глаз или волос. Большинство клеток в организме обычно содержат 46 хромосом, организованных в 23 пары. В каждой из этих 23 пар есть одна унаследованная хромосома от отца и одна — от матери.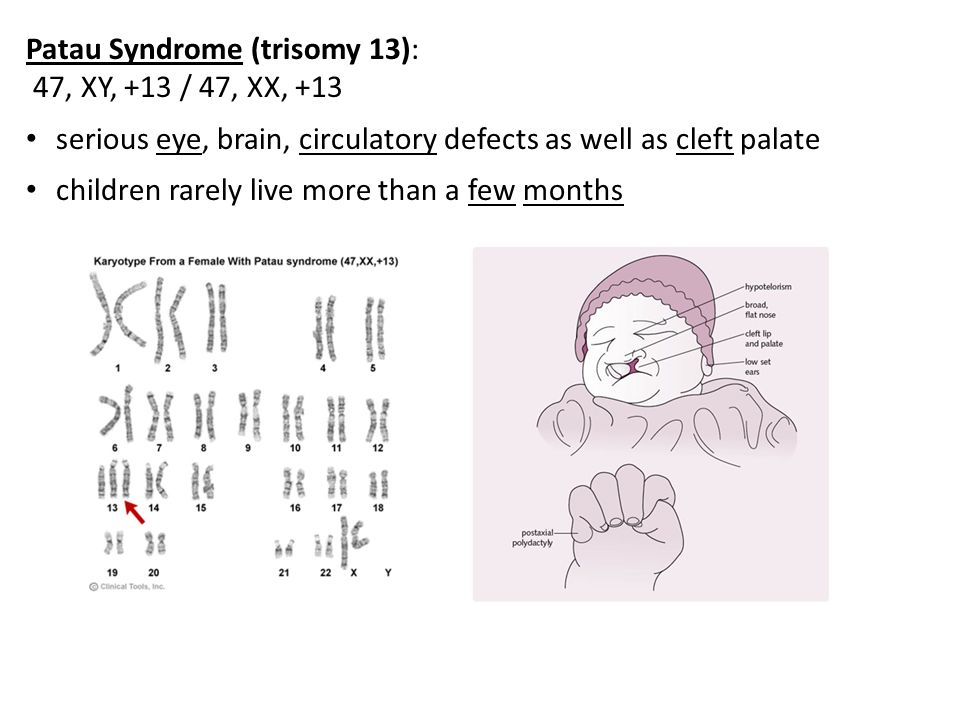 Из 23 пар 22 пары одинаковые у женских и мужских организмов, а одна оставшаяся определяет, являетесь вы мужчиной (XY) или женщиной (XX).
Из 23 пар 22 пары одинаковые у женских и мужских организмов, а одна оставшаяся определяет, являетесь вы мужчиной (XY) или женщиной (XX).
Мутации, из-за которых возникают наследственные заболевания, могут иметь доминантный или рецессивный характер наследования.
Доминантное наследование означает, что только одна копия гена — от матери или отца — должна иметь мутацию (или патогенный вариант гена) для проявления признака или заболевания. А при рецессивном типе человек наследует две измененные копии одного и того же гена.
Аутосомно-доминантный паттерн наследования
При аутосомно-доминантном наследовании заболеваний генетически обусловленная болезнь проявляется в том случае, если у человека есть хотя бы один мутированный ген, и этот ген не расположен на половых (Х и Y) хромосомах.
Болезнь Хантингтона и синдром Марфана — два примера аутосомно-доминантных болезней. Мутации в генах BRCA1 и BRCA2, которые также связаны с раком молочной железы, передаются по этой схеме.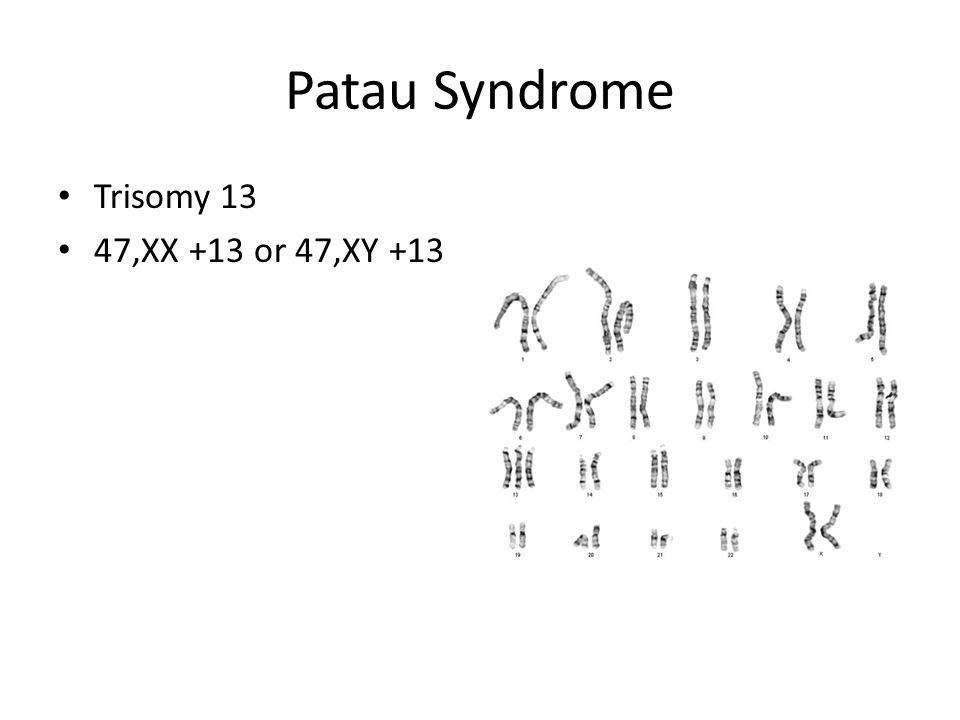
Аутосомно-рецессивный паттерн наследования
При аутосомно-рецессивном наследовании мутируют обе копии генов. Чтобы унаследовать аутосомно — рецессивное заболевание, такое как муковисцидоз, спинальная мышечная атрофия, или фенилкетонурия (ФКУ), оба родителя должны быть носителями. Ребенок наследует две копии дефектного гена — по одной от каждого родителя. Например, люди, имеющие одну копию гена с мутацией, а вторую — без мутации, называются носителями, потому что сами они здоровы.
Х-сцепленное рецессивное наследование
В Х-сцепленном рецессивном наследовании мутированный ген находится на Х-хромосоме. Болезнь проявляется только в случае, если другой Х-хромосомы с нормальной копией того же гена у человека нет.
Мышечная дистрофия Дюшенна, некоторые виды дальтонизма и гемофилия А — примеры рецессивных заболеваний, связанных с X-хромосомой. Мужчина с рецессивным заболеванием, связанным с X-хромосомой, передаст свою нетронутую Y-хромосому сыновьям, и ни один из них не пострадает. Если он передаст свою Х-хромосому (с дефектным геном) своим дочерям, то все они будут носителями болезни. У его дочерей может не быть симптомов или только легкие признаки заболевания, но они могут передать мутированный ген своим детям.
Если он передаст свою Х-хромосому (с дефектным геном) своим дочерям, то все они будут носителями болезни. У его дочерей может не быть симптомов или только легкие признаки заболевания, но они могут передать мутированный ген своим детям.
Женщины-носители рецессивного заболевания, связанного с X-хромосомой, часто имеют лёгкие признаки заболевания или вообще не имеют симптомов. Это связано с тем, что у женщин-носителей есть одна нормальная копия гена и одна мутированная копия. Нормальная копия обычно компенсирует дефектную копию в женском организме, в отличие от мужчин, у которых только одна X-хромосома.
Женщины, имеющие только один патологический ген, передают заболевание в среднем половине своих детей вне зависимости от пола. Женщины же, имеющие два патологических гена, передают заболевание всем своим детям. К таким заболеваниям относятся гемофилия А и дальтонизм.
Если вы знаете или предполагаете, что у вас или вашего партнера в семейной истории есть какое-либо генетическое заболевание, вы можете определить это с помощью Генетического теста Атлас. Генетическое консультирование поможет вам узнать о методах лечения, профилактических мерах и репродуктивных возможностях.
Генетическое консультирование поможет вам узнать о методах лечения, профилактических мерах и репродуктивных возможностях.
Как лечить наследственные заболевания и как с ними жить?
Раньше наследственные заболевания были неизлечимы. Сейчас это по-прежнему остаётся проблемой для многих заболеваний, но для некоторых из них методы лечения уже найдены. Например, это касается болезней, связанных с нарушением метаболизма.
При большинстве наследственных нарушений обмена веществ один фермент либо вообще не вырабатывается организмом, либо вырабатывается в форме, которая не работает. Например, при отсутствии какого-либо фермента в организме могут накапливаться токсичные вещества или может не синтезироваться необходимый продукт — как при гемохроматозе 1 типа.
При этом заболевании организм поглощает слишком много железа из пищи и не может естественным образом избавиться от избытка. Это может привести к чрезмерному накоплению железа в сердце, поджелудочной железе и печени.
Лечение генетических нарушений обмена веществ следует двум общим принципам:
- Необходимо сократить или исключить прием любой пищи или лекарств, которые не усваиваются организмом.
- Заменить или восполнить отсутствующий или неактивный фермент для восстановления метаболизма с помощью диеты и/или лекарств.
Есть более серьезные и распространенные наследственные заболевания, которые не лечатся. Например, мековисцидоз — скопление слизи в лёгких и в пищеварительной системе. От муковисцидоза нет лекарства, но разные методы контроля симптомов помогают предотвращать или уменьшать осложнения и облегчать жизнь с этим заболеванием.
Со временем муковисцидоз прогрессирует и может привести к летальному исходу, особенно при наличии сопутствующих инфекций. Сегодня благодаря достижениям медицины около половины людей с муковисцидозом доживают до 40 лет. Дети, рожденные с этим заболеванием в наши дни, смогут прожить ещё дольше.
Одно из самых тяжелых наследственных заболеваний, спинальная мышечная атрофия, также с недавнего времени поддается лечению с помощью генной терапии. Но доступен этот метод далеко не каждому. Препарат для лечения СМА — самый дорогой лекарственный препарат в мире.
Лечение или купирование генетических заболеваний стало возможным благодаря международному проекту «Геном человека» по изучению и картированию генов человека, произошел прорыв в диагностике и лечении наследственных заболеваний. Результаты проекта помогают не только находить гены, мутации в которых приводят к заболеваниям, но и диагностировать их с максимальной точностью.
Как я могу узнать, что являюсь носителем генетического заболевания?
Наши гены содержат инструкции, которые сообщают организму, как правильно функционировать. При изменении этих инструкций развиваются различные заболевания. Во многих случаях симптомы впервые проявляются в зрелом возрасте, поэтому иногда мы не знаем, что являемся носителями. Предупредить риски развития и передачи наследственного заболевания можно с помощью Генетического теста Атлас.
На заметку:
- Наследственные заболевания — это заболевания, обусловленные генными или хромосомными мутациями.
- При совпадении у партнеров статусов носительства определенных болезней есть высокий риск рождения ребенка с наследственным заболеванием. Поэтому при планировании беременности важно пройти генетическое тестирование.
- Мутации, из-за которых возникают наследственные заболевания, могут иметь доминантный или рецессивный характер наследования. При доминантном наследовании только одна копия гена — от матери или отца — должна иметь мутацию для проявления признака или заболевания. А при рецессивном типе человек наследует две измененные копии одного и того же гена.
- Большинство наследственных заболеваний неизлечимы. Течение некоторых из них можно контролировать с помощью лекарств и диеты.
- Определить наличие и риск развития наследственного заболевания можно с помощью Генетического теста Атлас.
 Поэтому при планировании беременности важно пройти генетическое тестирование.
Поэтому при планировании беременности важно пройти генетическое тестирование.Трисомия 13 — Министерство здравоохранения Миннесоты
Для поставщиков: Эта информация соответствует требованиям Закона об осведомленности о пренатальной трисомии (MS 145.471), который требует предоставления учебных материалов беременным женщинам после положительного скринингового теста на трисомию 13 (синдром Патау). Приемлемую альтернативу этому информационному листку под названием «Общие сведения о пренатальном скрининге и диагностике» можно получить в Национальном центре пренатальных и послеродовых ресурсов Lettercase.Эта информация доступна в Интернете через веб-приложение или путем запроса бесплатных печатных материалов на их веб-сайте. Приемлемую альтернативу этому информационному листку под названием «Общие сведения о пренатальном скрининге и диагностике» можно получить в Национальном центре пренатальных и послеродовых ресурсов Lettercase.Эта информация доступна в Интернете через веб-приложение или путем запроса бесплатных печатных материалов на их веб-сайте. |
Описание состояния
Трисомия 13, также называемая синдромом Патау, представляет собой тяжелое хромосомное заболевание с множественными пороками развития из-за дополнительной копии всей или части хромосомы 13. Причина этой дополнительной копии хромосомы 13 неизвестна. Младенцы обычно идентифицируются при рождении по нескольким узнаваемым физическим особенностям, но диагноз подтверждается генетическим тестированием.Люди с трисомией 13 часто имеют пороки сердца, аномалии головного или спинного мозга. Младенцы могут быть маленькими при рождении и иметь характерный внешний вид лица вместе с плохим мышечным тонусом.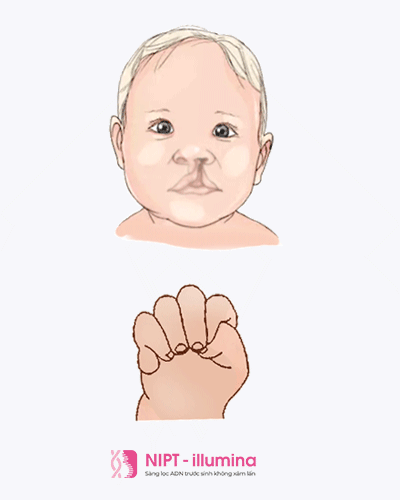 Некоторые из этих особенностей включают: небольшой размер головы (микроцефалия), маленькие глаза (микрофтальм), расщелина губы / неба и вариации формы ушей.
Некоторые из этих особенностей включают: небольшой размер головы (микроцефалия), маленькие глаза (микрофтальм), расщелина губы / неба и вариации формы ушей.
Распространенность
Трисомия 13 встречается у 1 из 10 000–16 000 родов, и заболеваемость увеличивается с увеличением возраста матери. Риск рецидива при будущих беременностях составляет 1%.Большинство случаев не передаются по наследству и возникают в результате случайного образования яйцеклеток и сперматозоидов у здоровых родителей.
Наша программа отслеживает трисомию 13 среди живорожденных в отдельных округах с 2005 года и постепенно расширяется по всему штату.
- Используя данные о рождении жителей округа Хеннепин и Рэмси в период с 2012 по 2016 год, мы обнаружили, что менее 1 ребенка родился с трисомией 13 на 10 000 рождений.
- Используя эти данные, мы оцениваем, что в Миннесоте ежегодно рождается около 6 детей с трисомией 13.
Общие ассоциированные состояния
Младенцы с трисомией 13 будут иметь характерную группу проблем, которая может включать следующее: микроцефалия (маленький размер головы), заячья губа и / или расщелина неба (дефект лица и / или рта), омфалоцеле (дефект брюшной полости), расщелина позвоночника ( открытый дефект позвоночника), микрофтальм (маленькие глаза), анофтальм (отсутствие глаз), дефекты кожи головы, полидактилия (лишние пальцы рук и ног), крипторхизм (неопущение яичек), омфалоцеле (кишечник ребенка, печень или другие органы выходят за пределы живота), голопрозэнцефалия (анатомический дефект головного мозга с вовлечением переднего мозга), дефекты почек и кожные дефекты волосистой части головы. Около 80% младенцев с трисомией 13 также имеют порок сердца. Тип порока сердца различается, но чаще всего это дефект межжелудочковой перегородки (отверстие между нижними камерами сердца), дефект межпредсердной перегородки (отверстие между верхними камерами сердца), открытый артериоз протока (кровеносный сосуд из зародышевого периода, который не закрывается) или декстрокардия, при которой сердце располагается с правой стороны грудной клетки, а не с левой.
Около 80% младенцев с трисомией 13 также имеют порок сердца. Тип порока сердца различается, но чаще всего это дефект межжелудочковой перегородки (отверстие между нижними камерами сердца), дефект межпредсердной перегородки (отверстие между верхними камерами сердца), открытый артериоз протока (кровеносный сосуд из зародышевого периода, который не закрывается) или декстрокардия, при которой сердце располагается с правой стороны грудной клетки, а не с левой.
Краткосрочное лечение и результаты
Родители ребенка с трисомией 13 сталкиваются со многими трудными решениями относительно ухода за своим ребенком.Младенцам будет обеспечен комфортный уход, и 80% из них не доживут до первого месяца жизни. Большинство из них не доживут до первой недели, несмотря на медицинское вмешательство. Несмотря на хорошее потребление калорий, многие дети с хромосомными дефектами демонстрируют медленный рост. Родителям будет предложено генетическое тестирование и консультация.
Некоторые дети страдают менее серьезно и преодолеют многие трудности; таким младенцам следует регулярно посещать детский сад для вакцинации и упреждающего руководства, с частыми проверками на предмет нарушений зрения и слуха, сколиоза, задержки развития и симптомов других состояний, которые можно лечить.
Долгосрочное лечение и результаты
Практически все 5% детей, выживших после первого года жизни, будут демонстрировать задержку в развитии и росте. Программы раннего вмешательства и специальное обучение будут очень важны для относительно небольшого числа детей с трисомией 13, переживших трудные первые месяцы жизни.
Распространенные осложнения
Список возможных осложнений очень велик, потому что состояние трисомии 13 поражает очень много систем организма.Инфекция и трудности с кормлением будут серьезными проблемами при уходе за этими младенцами.
Влияние на развитие детей
Рождение ребенка с ослабленным соматическим здоровьем является трудным, и семьи найдут поддержку в других семьях, которые столкнулись с подобными обстоятельствами. Родители часто учатся от других родителей, как защищать потребности своего ребенка. Группы поддержки можно найти в Интернете, а также во многих сообществах для решения многих повседневных проблем родителей. Принятие решений для детей с хромосомными дефектами сложно и сложно, и родители нуждаются в информации и поддержке со стороны их медицинских работников и сообщества. Ниже перечислены организации, которые предоставляют практическую информацию и поддержку семьям с ребенком с трисомией 18.
Принятие решений для детей с хромосомными дефектами сложно и сложно, и родители нуждаются в информации и поддержке со стороны их медицинских работников и сообщества. Ниже перечислены организации, которые предоставляют практическую информацию и поддержку семьям с ребенком с трисомией 18.
Симптомы синдрома Патау, Лаборатория тестирования синдрома Патау, Кариотип синдрома Патау
Показания для синдрома Патау (трисомия 13)
Синдром Патау, также известный как трисомия 13, представляет собой синдром, при котором у пациента есть дополнительная хромосома 13, симптомы синдрома Патау
включают:
Умственная и моторная отсталость
Полидактилия (дополнительные пальцы)
Голопрозэнцефалия (нарушение правильного разделения переднего мозга).
Пороки сердца
Структурные дефекты глаза, включая микрофтальмию, аномалию Петерса, катаракту, радужную оболочку и / или глазное дно (колобому), дисплазию сетчатки или отслоение сетчатки, сенсорный нистагм, корковую потерю зрения и гипоплазию зрительного нерва
Менингомиелоцеле (дефект позвоночника)
Омфалоцеле (абдоминальный дефект)
Аномальные гениталии
Аномальный рисунок ладони
Наложение пальцев на большой палец.
Аплазия кутиса (отсутствующая часть кожи / волос)
Выступающая пятка
Микроцефалия
Низко посаженные уши
Расщелина неба или заячьей губы
Процедура сбора
• Амниотическая жидкость
Откажитесь от первых 2 см3 жидкости, чтобы снизить риск заражения материнскими клетками.С соблюдением правил асептики перенесите 15–20 куб. См в стерильные пластиковые конические пробирки с указанием имени и даты рождения пациента.
• CVS
В асептических условиях перенесите 5–10 мг в стерильные пластиковые конические пробирки, содержащие стерильную транспортную среду. Наклейте на пробирку имя и дату рождения пациента.
• Периферическая кровь
Соберите 5–10 куб. См цельной крови в зеленую верхнюю пробирку (гепарин натрия). Наклейте на пробирку имя и дату рождения пациента.
Заполненная форма заявки на тестирование должна быть приложена к каждому образцу.Транспортный комплект предоставляется по запросу.
Требования к образцам
Лабораторное тестирование на синдром Патау может быть выполнено как 24-часовое FISH-исследование SAT на интерфазных клетках с последующим полным цитогенетическим анализом (кариотип)
Температура транспортировки
Комнатная температура (НЕ замораживать, не замораживать и не центрифугировать)
Методология
24-часовое лабораторное тестирование на синдром Патау (трисомия 13) может быть выполнено путем гибридизации Fluorescence in situ на интерфазных ядрах с последующим подтверждающим цитогенетическим анализом.
Причины отказа
Замороженный образец, неподходящий контейнер
Срок выполнения
FISH: 24 часа
Цитогенетический анализ: 7-10 дней
NDIS Тип инвалидности: синдром Патау (трисомия 13)
- Просмотров: 2616
Этот информационный бюллетень содержит информацию о синдроме Патау, его частоте, признаках и симптомах, скрининге и лечении.
Синдром Патау, также известный как трисомия 13, является генетическим заболеванием, при котором имеется дополнительная копия хромосомы 13.
Синдром характеризуется тяжелой умственной отсталостью и связанными с ней заболеваниями, такими как пороки сердца и аномалии головного и спинного мозга.
Из-за наличия этих опасных для жизни заболеваний многие новорожденные с синдромом Патау умирают в течение первой недели жизни, причем примерно от 5 до 10% детей живут старше 12 месяцев.
Это не наследственное заболевание, а скорее результат случайных событий во время образования яйцеклеток и сперматозоидов у здоровых родителей.
Частота
Синдром Патау встречается примерно у 1 из 16 000 новорожденных
Признаки и симптомы
- Затруднения при кормлении, ведущие к плохому приросту и весу
- Тяжелая умственная отсталость
- Черты лица могут включать широко расставленные глаза с вертикальными кожными складками на внутренних углах глаза, широкий плоский нос и расщелину неба или губы.
- Пороки сердца
- Патологии головного и спинного мозга
- Плохое развитие глаз
- Дополнительные пальцы рук или ног
- Расщелина неба (отверстие в небе)
- Слабость мышц
- Патология почек
- Неопустившиеся яички у мальчиков
Просеивание
Антенатальные скрининговые тесты на трисомию 13 проводятся в сочетании со скринингом на синдром Дауна в первом триместре.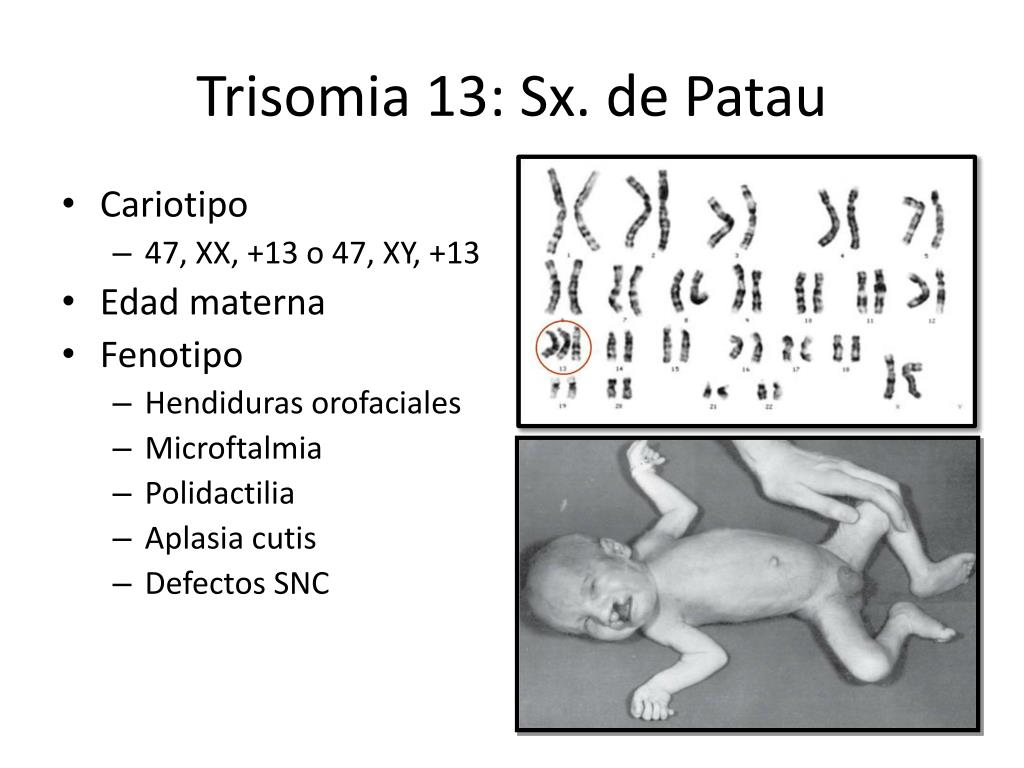 Это делается с помощью анализа крови и ультразвука. Поэтому диагноз обычно ставится до рождения ребенка.
Это делается с помощью анализа крови и ультразвука. Поэтому диагноз обычно ставится до рождения ребенка.
Лечение
В настоящее время не существует методов лечения синдрома Патау. Новорожденные после рождения помещаются в реанимацию.
Список литературы
Синдром Патау (2019). Получено с https://ghr.nlm.nih.gov/condition/trisomy-13#diagnosis
.Синдром Патау (2019). Получено с http://brochures.mater.org.au/brochures/mater-mothers-hospital/patau-syndrome-trisomy-13
.Синдром Патау Определение и примеры
Определение
существительное
Генетическое заболевание, вызванное генетическими изменениями в хромосоме 13, такими как дополнительная копия хромосомы 13 (трисомия) или транслокация части хромосомы 13
Дополнение
Синдром Патау — одно из различных генетических заболеваний человека, вызванных наличием дополнительного генетического материала в клетках организма. При синдроме Патау дополнительный генетический материал представляет собой дополнительную копию всей или частичной части хромосомы 13. Хромосома 13 — это хромосома, которая может содержать около 300 или менее 400 генов, с центромерой, расположенной ближе к концу ( акроцентрический). Это аутосома, которая обычно встречается в виде двух копий, то есть одна происходит от матери, а другая от отца. Есть случаи, когда имеется избыток хромосомы 13. Хромосомная дупликация и нерасхождение во время мейоза являются возможными причинами, которые приводят к появлению дополнительного генетического материала.Наличие трех копий вместо двух называется трисомией. Таким образом, синдром Патау также называют трисомией 13. Другой возможной причиной синдрома Патау является транслокация, при которой часть хромосомы, такая как хромосома 13, перемещается не на место или перемещается на другую хромосому во время эмбрионального развития. Некоторые случаи синдрома Патау относятся к мозаичному типу, когда поражаются только некоторые клетки тела.
При синдроме Патау дополнительный генетический материал представляет собой дополнительную копию всей или частичной части хромосомы 13. Хромосома 13 — это хромосома, которая может содержать около 300 или менее 400 генов, с центромерой, расположенной ближе к концу ( акроцентрический). Это аутосома, которая обычно встречается в виде двух копий, то есть одна происходит от матери, а другая от отца. Есть случаи, когда имеется избыток хромосомы 13. Хромосомная дупликация и нерасхождение во время мейоза являются возможными причинами, которые приводят к появлению дополнительного генетического материала.Наличие трех копий вместо двух называется трисомией. Таким образом, синдром Патау также называют трисомией 13. Другой возможной причиной синдрома Патау является транслокация, при которой часть хромосомы, такая как хромосома 13, перемещается не на место или перемещается на другую хромосому во время эмбрионального развития. Некоторые случаи синдрома Патау относятся к мозаичному типу, когда поражаются только некоторые клетки тела.
Симптомы синдрома Патау могут различаться в зависимости от степени тяжести. Некоторые из симптомов — умственная отсталость, голопроэнцефалия, менингомиелоцеле, пороки сердца, покатый лоб, микроцефалия, структурные дефекты глаз, заячья губа, низко посаженные уши, полидактилия, качающиеся ступни, аномальные гениталии и т. Д.
Синдром Патау назван в честь Клауса Патау, американского генетика немецкого происхождения, который сообщил о синдроме и связал его с трисомией в 1960 году.
Синоним (ы):
См. Также:
Последнее обновление 28 июля 2021 г.
Синдром Патау: информация для родителей
Public Health England ( PHE ) создал эту информацию от имени NHS. В нем слово «мы» относится к службе NHS, которая проводит обследование.
Вы читаете эту информацию, потому что после 20-недельного сканирования у вашего ребенка подозревается синдром Патау (также известный как трисомия 13 или T13).
Эта информация должна помочь вам и вашим специалистам в области здравоохранения обсудить следующие этапы ухода за вами и вашим ребенком. Он должен поддерживать, но не заменять обсуждения, которые вы ведете со специалистами в области здравоохранения.
Обнаружение проблемы с развитием ребенка может вызывать беспокойство. Важно помнить, что вы не одиноки.
Мы направим вас к специалистам, которые:
- предоставит более точную информацию о состоянии вашего ребенка
- ответьте на ваши вопросы
- поможет вам спланировать следующие шаги
О синдроме Патау
Внутри клеток нашего тела есть крошечные структуры, называемые хромосомами. Эти хромосомы несут гены, определяющие наше развитие. Клетки человеческого тела содержат 46 хромосом. Изменения, происходящие в сперматозоидах или яйцеклетках, могут привести к появлению у ребенка лишних хромосом.
У младенцев с синдромом Патау есть дополнительная копия хромосомы 13 во всех или некоторых клетках.
Существует 3 типа синдрома Патау: полный, мозаичный и частичный синдром Патау. Насколько серьезным является заболевание, обычно зависит от типа синдрома Патау у вашего ребенка. Скрининг на 20-недельном сканировании не может сказать вам, какой тип синдрома Патау может быть у вашего ребенка.
Во многих случаях синдром Патау — это состояние, ограничивающее жизнь, и показатели выживаемости низкие. Вылечить это состояние невозможно.
Все дети, рожденные с синдромом Патау, будут иметь проблемы с обучением и будут иметь широкий спектр проблем со здоровьем, некоторые из которых могут быть чрезвычайно серьезными. У них могут быть проблемы с их:
- сердце
- дыхательная система
- почки
- пищеварительная система
Младенцы, рожденные с полным синдромом Патау, могут медленно прогрессировать в своем развитии, несмотря на свои сложные потребности.
Младенцы, рожденные с мозаичным или частичным синдромом Патау, могут иметь менее серьезные проблемы со здоровьем, но невозможно узнать об этом до рождения ребенка.
Причины
Мы не знаем точно, что вызывает синдром Патау. Это не вызвано тем, что вы сделали или не сделали. Младенцы с синдромом Патау рождаются у матерей любого возраста, но шанс зачать ребенка с этим заболеванием увеличивается с возрастом матери.
Вы сможете обсудить ваши индивидуальные обстоятельства со специалистами.
Синдром Патау встречается примерно у одного ребенка из 4000 (0,03%).
Как мы обнаруживаем синдром Патау
Мы проводим скрининг на синдром Патау при «20-недельном сканировании» (между 18 +0 и 20 +6 неделями беременности).Скрининг на синдром Патау также является частью комбинированного теста, предлагаемого на ранних сроках беременности между 10 и 14 неделями.
Контрольные обследования и записи на прием
Поскольку результат сканирования предполагает, что у вашего ребенка может быть такое заболевание, как синдром Патау, мы направляем вас в группу специалистов, которые заботятся о беременных женщинах и их младенцах до их рождения. Они могут находиться в больнице, в которой вы в настоящее время получаете дородовую помощь, или в другой больнице.Команда специалистов может предложить вам дополнительные тесты, такие как пробы ворсинок хориона ( CVS ) или амниоцентез, которые смогут подтвердить, есть ли у вашего ребенка синдром Патау и что это может означать.
Они могут находиться в больнице, в которой вы в настоящее время получаете дородовую помощь, или в другой больнице.Команда специалистов может предложить вам дополнительные тесты, такие как пробы ворсинок хориона ( CVS ) или амниоцентез, которые смогут подтвердить, есть ли у вашего ребенка синдром Патау и что это может означать.
Может быть полезно записать любые вопросы, которые вы хотите задать, до того, как вы посетите группу специалистов.
Результат
Нет лекарства от синдрома Патау. К сожалению, у многих детей с синдромом Патау происходит выкидыш во время беременности. Из тех младенцев, родившихся живыми, около 11% доживают до своего первого дня рождения.Некоторые дети могут дожить до взрослого возраста, но это бывает редко.
Ожидаемая продолжительность жизни детей, рожденных с мозаичным или частичным типом синдрома Патау, может быть гораздо более изменчивой.
Младенцам с синдромом Патау после рождения может потребоваться специализированная помощь и лечение. Это будет сосредоточено на симптомах состояния, которое у них есть.
Это будет сосредоточено на симптомах состояния, которое у них есть.
Примерно у половины детей с синдромом Патау есть заячья губа и нёбо. Младенцы с синдромом Патау также могут иметь низкий вес при рождении.
Следующие шаги и варианты выбора
Если будет подтверждено, что у вашего ребенка синдром Патау, вы можете поговорить с командой, осуществляющей уход за вами во время беременности, о состоянии вашего ребенка и возможных вариантах. Это будет включать продолжение беременности или прерывание беременности. Возможно, вам захочется узнать больше о синдроме Патау. Может быть полезно поговорить с организацией поддержки, имеющей опыт помощи родителям в этой ситуации.
Если вы решите продолжить беременность, команда специалистов поможет вам спланировать лечение.Команда обсудит с вами, как вы хотите, чтобы о вашем ребенке заботились после рождения. В зависимости от конкретных симптомов вашего ребенка может быть предложена паллиативная помощь. Паллиативная помощь детям направлена на обеспечение максимально возможного качества жизни и заботы о каждом ребенке с ограничивающим жизнь заболевании и его семье.
Если вы решите прервать беременность, вам будет предоставлена информация о том, что это влечет за собой и как вам будет оказана поддержка. Вам должен быть предложен выбор, где и как прервать беременность, а также поддержка, индивидуальная для вас и вашей семьи.
Только вы знаете, какое решение будет лучшим для вас и вашей семьи.
Какое бы решение вы ни приняли, медицинские работники вас поддержат.
Будущие беременности
Если вы решите завести еще одного ребенка, у него вряд ли будет синдром Патау.
Младенцы с синдромом Патау рождаются у матерей любого возраста, но вероятность этого увеличивается по мере взросления матери.
Вас могут направить к генетическому консультанту, чтобы обсудить будущую беременность.
Дополнительная информация и поддержка
Antenatal Results and Choices ( ARC ) — это национальная благотворительная организация, которая поддерживает людей, принимающих решения о скрининге и диагностике, а также о продолжении беременности.
Организация поддержки трисомии 13/18 ( SOFT UK ) — это национальная благотворительная организация, которая поддерживает семьи, страдающие синдромом Патау, синдромом Эдварда и связанными с ним заболеваниями.
Дополнительную информацию о синдроме Патау можно найти на веб-сайте NHS.
Бенефтис для синдрома Патау
В 2010 году Управление социального обеспечения получило более трех миллионов заявлений на получение пособия по социальному страхованию по инвалидности. Более двух третей этих заявок были отклонены на начальном этапе процесса подачи заявок. Хотя многие из этих исков были поданы работниками-инвалидами, которые больше не могли поддерживать работу из-за их инвалидности, некоторые из них были поданы родителями в отношении детей, у которых были диагностированы тяжелые изнурительные заболевания.
Когда у ребенка диагностируется серьезное и опасное для жизни заболевание, финансовые последствия могут быть огромными. В некоторых случаях пособия по социальному обеспечению по инвалидности могут помочь частично компенсировать финансовое бремя, вызванное болезнью ребенка. К счастью, многие из родителей, которые подают заявления на получение пособия по социальному страхованию по инвалидности для ребенка-инвалида, могут избежать сложностей и стресса, связанных со стандартным процессом подачи заявления на социальное обеспечение по инвалидности.
В 2008 году Управление социального обеспечения ввело программу социальных пособий.Согласно руководству по выплате пособий по состраданию, некоторые заявители могут получить пособие по инвалидности в течение нескольких недель, вместо того, чтобы ждать месяцами или даже годами утверждения об инвалидности. Существует 88 условий, позволяющих заявителю обрабатывать претензию в соответствии с этими руководящими принципами. Среди них синдром Патау.
Если вашему ребенку был поставлен диагноз синдрома Патау, следующая информация поможет вам понять процесс подачи заявления о потере трудоспособности и то, что вы можете сделать, чтобы повысить свои шансы на получение быстрого одобрения пособий по инвалидности для вашего ребенка в соответствии с руководящими принципами SSA по выплате сострадательных пособий.
Синдром Патау (трисомия 13) — состояние и симптомы
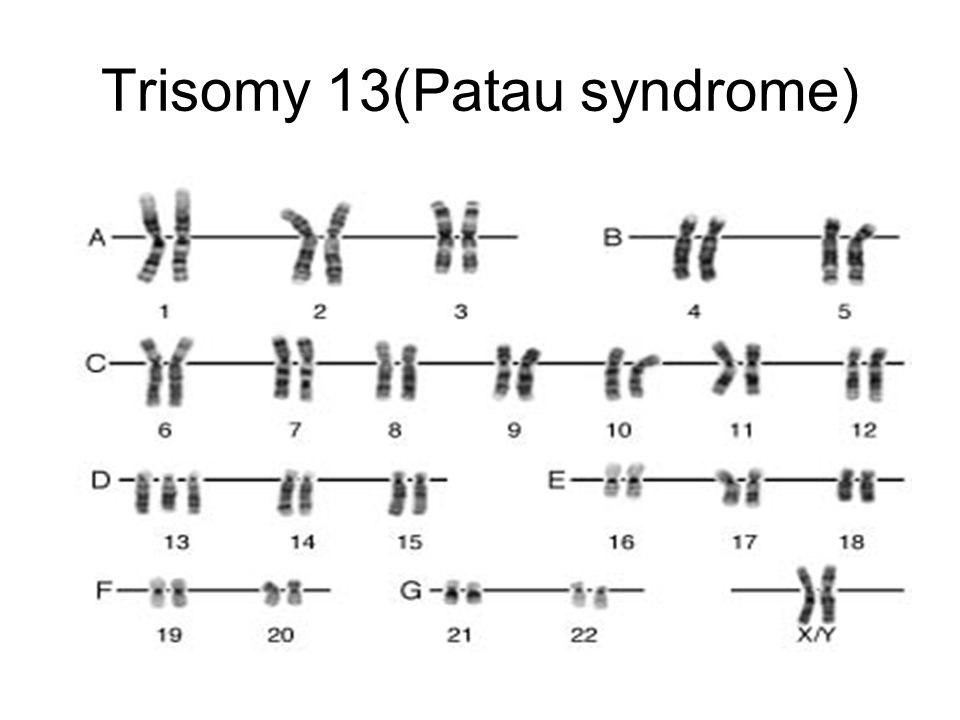
Синдром Патау, также известный как трисомия 13, является редким генетическим заболеванием, в результате которого рождается ребенок с тремя копиями генетического материала, содержащегося в тринадцатой хромосоме. Заболевание поражает примерно одного из 10 000 новорожденных. У здоровых младенцев есть только две копии этого генетического материала. Для детей, рожденных с трисомией 13, дополнительный материал влияет на развитие больного ребенка.
Существует три различных типа трисомии 13, включая случаи трисомии 13, когда все клетки содержат дополнительный генетический материал, трисомию 13, когда некоторые клетки содержат дополнительную хромосому, и частичную трисомию, когда в клетках присутствует только часть дополнительной хромосомы. . Тяжесть состояния будет зависеть от типа трисомии 13, которым страдает ребенок.
В отличие от многих инвалидизирующих состояний новорожденных, синдром Патау не передается от родителей к ребенку.Вместо этого это состояние возникает в результате дефекта яйцеклетки или сперматозоидов, которые сформировали плод. Симптомы синдрома Патау могут варьироваться в зависимости от тяжести состояния и от того, были ли затронуты все или только некоторые клетки. Общие симптомы трисомии 13 включают заячью губу, волчью пасть, сжатые руки, близко посаженные глаза, нарушение мышечного тонуса, лишние пальцы рук или ног, грыжу, низко посаженные уши, нарушение умственного развития, отсутствие кожи, судороги, деформации скелета, маленькие глаза , маленькая голова, маленькая нижняя челюсть и неопущенные яички.
Стандартного курса лечения младенцев, рожденных с синдромом Патау, не существует. Вместо этого лечение является специализированным и ориентировано на симптомы каждого отдельного пациента. Нередко дети, рожденные с этим заболеванием, нуждаются в хирургическом вмешательстве, физиотерапии, логопеде, трудотерапии и других поддерживающих лечебных мероприятиях.
Ожидаемая продолжительность жизни ребенка, рожденного с синдромом Патау, будет варьироваться в зависимости от конкретных симптомов и тяжести состояния.Во многих случаях ребенок, рожденный с синдромом Патау, не доживает до первого месяца жизни.
Подача заявки на социальное обеспечение по инвалидности с синдромом Патау (трисомия 13)
Рождение ребенка с диагнозом синдрома Патау может быть эмоционально разрушительным. Многие родители задаются вопросом, как они будут удовлетворять потребности ребенка, сохраняя при этом финансовые потребности семьи. Во многих случаях могут помочь пособия по социальному обеспечению по инвалидности.
К счастью, синдром Патау является одним из 88 условий, при которых заявитель по инвалидности может подавать заявление в соответствии с рекомендациями SSA по выплате пособий по состраданию.Это означает, что ваш ребенок может быть одобрен для получения пособия по инвалидности в течение нескольких недель, вместо того, чтобы месяцами ждать завершения стандартного процесса подачи заявления о нетрудоспособности.
При подаче заявления по социальному страхованию на основании диагноза синдрома Патау убедитесь, что вы подробно ответили на все поставленные вам вопросы и правильно заполнили заявление. Неправильно оформленные заявления по инвалидности являются одной из основных причин отказа в выплате пособия по инвалидности. Вы также должны обязательно приложить к заявлению об инвалидности полную копию медицинских карт вашего ребенка, включая результаты анализов и письменные заявления лечащих врачей.Эти документы потребуются для своевременного утверждения пособий по социальному страхованию вашего ребенка по инвалидности. Регулярно проверяйте статус вашего заявления и при необходимости подайте апелляцию.
Ваш синдром Патау (трисомия 13) Случай инвалидности по социальному обеспечению
Многие из родителей, которые подают заявления на получение пособия по социальному обеспечению по инвалидности для детей, у которых было диагностировано состояние, подпадающее под списки пособий по состраданию SSA, предполагают, что заявление их ребенка будет автоматически одобрено Управлением социального обеспечения.Это не обязательно так. Вам все равно нужно будет убедиться, что ваше заявление заполнено должным образом и предоставлены достаточные медицинские доказательства для получения пособия по инвалидности в связи с заболеванием вашего ребенка.
Если вы хотите повысить свои шансы на получение ускоренного одобрения заявления вашего ребенка об инвалидности, вам следует подумать о том, чтобы воспользоваться услугами квалифицированного юриста или адвоката по инвалидности. Эти специалисты могут помочь вам в подготовке вашего заявления о потере трудоспособности по социальному обеспечению и обеспечат представление вашего заявления в Администрацию социального обеспечения в наилучшем свете.
Синдром Патау: факты и информация
Опубликовано: 16.02.2010 — Обновлено: 14.01.2016
Автор: Disabled World | Контакты: Disabled World (Disabled-World.com)
Синопсис: Синдром Патау — генетическое заболевание, при котором тринадцатая хромосома человека появляется трижды вместо двух.
Main Digest
Определение синдрома Патау
Синдром Патау, также называемый «трисомией 13», представляет собой форму генетического нарушения, при котором вся или часть тринадцатой хромосомы человека появляется трижды вместо двух в клетках их тела.У некоторых людей, страдающих этим синдромом, только процент клеток может содержать дополнительную тринадцатую хромосому, называемую «мозаицизмом», тогда как дополнительные клетки действительно содержат среднюю пару хромосом. Лишний материал мешает нормальному процессу развития человека, что приводит к серьезной умственной отсталости и физическим отклонениям в ряде частей его тела.
Синдром, вызванный хромосомной аномалией, при которой некоторые или все клетки тела содержат дополнительный генетический материал из хромосомы 13.Это может происходить либо из-за того, что каждая клетка содержит полную дополнительную копию хромосомы 13 (нарушение, известное как трисомия 13 или трисомия D), либо из-за того, что каждая клетка содержит дополнительную частичную копию хромосомы (например, Робертсоновская транслокация), либо из-за мозаичного Патау. синдром.
Синдром Патау встречается примерно у одного из десяти — шестнадцати тысяч младенцев. Большинство людей с синдромом Патау не унаследовали его, а получили синдром в результате случайных событий во время образования яйцеклеток и сперматозоидов у здоровых родителей.Из-за наличия ряда опасных для жизни медицинских проблем многие дети с синдромом Патау умирают в первые несколько дней или недель своей жизни. От пяти до десяти процентов детей с синдромом Патау доживают до первого года жизни.
Младенцы, рожденные с синдромом Патау, обладают узнаваемым паттерном физических особенностей, который во многих случаях позволяет медицинским работникам поставить диагноз синдрома. Заметные физические врожденные дефекты, а иногда и анатомические изменения внутренних органов ребенка.Важные результаты включают небольшой размер головы, называемый «микроцефалией», маленькие глаза или «микрофтальмия», а иногда и отсутствие глаза или дефект в развитии сетчатки у ребенка. Примерно у шестидесяти процентов детей с синдромом Патау имеется волчья пасть или заячья губа. Дополнительные результаты могут включать изменения формы ушей ребенка, лишних пальцев на ногах или пальцах, а также изменения в ладони руки ребенка. Изменения могут происходить в развитии пятки или стопы ребенка, и они могут проявляться в виде «качательной стопы».Дополнительные названия для синдрома Патау включают:
- Трисома
- Трисомия
- Трисоми
- Трисомия 13
- Фетальная анеуплоидия
- Синдром Патау
- Синдром трисомии D
- Синдром трисомии 13
Синдром трисомии 13
У людей, страдающих синдромом Патау, часто бывают пороки сердца, маленькие или плохо развитые глаза, аномалии спинного или головного мозга, расщелина губы или неба, лишние пальцы ног или пальцев, а также снижение мышечного тонуса.Множество детей с синдромом Патау не растут и не набирают вес с ожидаемой скоростью, испытывают трудности с кормлением, а также имеют эпизоды, при которых у них временно прекращается спонтанное дыхание или «апноэ». Дополнительные признаки синдрома Патау могут включать:
- Грыжи
- Припадки
- Маленькие глаза
- Маленькая голова
- Низко посаженные уши
- Дефекты кожи головы
- Сжатые руки
- Маленькая нижняя челюсть
- Умственная отсталость
- Одинарная ладонная складка
- Неопущенное яичко
- Скелетные аномалии
- Отверстие, расщепление или расщелина радужной оболочки
- Близко посаженные глаза — глаза могут срастаться вместе
Примерно у восьмидесяти процентов детей с синдромом Патау будет врожденный порок сердца .Формы пороков сердца, которые могут возникнуть у детей с этим синдромом, могут включать дефект межжелудочковой перегородки, который затрагивает нижние камеры сердца и препятствует правильному перекачиванию крови. У них может наблюдаться дефект межпредсердной перегородки и разрыв между двумя верхними камерами сердца, что затрудняет перекачку сердцу достаточного количества богатой кислородом крови к тканям тела. «Открытый артериоз протока» — это еще одна форма порока сердца, с которой могут столкнуться дети с синдромом Патуа. Он включает дефект, при котором канал обычно закрывается незадолго до рождения.Они также могут испытывать декстрокардию, расположение сердца ребенка на правой стороне груди или, в некоторых случаях, более серьезные пороки сердца. Общие и важные врожденные дефекты, связанные с синдромом Патау, включают:
- Омфалоцеле
- Дефекты почек
- Голопрозэнцефалия
- Кожные дефекты волосистой части головы
Общие расстройства как у младенцев, так и у маленьких детей с синдромом Патау включают трудности с кормлением, медленный пост натальный рост, гастроэзофагеальный рефлюкс, судороги, апноэ, дефекты почек и гипертония.Младенцы и дети также могут страдать сколиозом и отклонениями в развитии.
Причины синдрома Патау
Синдром Патау возникает, когда дополнительная ДНК из тринадцатой хромосомы появляется в некоторых или во всех клетках тела человека. «Мозаицизм» — это состояние, при котором наличие дополнительной тринадцатой хромосомы присутствует только в некоторых клетках человека. А, «частичная трисомия», возникает, когда в клетках человека присутствует часть дополнительной тринадцатой хромосомы.Лишние материалы мешают нормальному развитию человека. Большинство людей с синдромом Патау не унаследовали этот синдром. События, которые привели к синдрому, произошли либо в сперме, либо в яйцеклетке, из которой сформировался плод.
Большинство людей с синдромом Патау имеют три копии тринадцатой хромосомы в каждой клетке своего тела вместо обычных двух копий. Избыточный генетический материал нарушает ход их развития, что приводит к характерным чертам синдрома Патау.Физические особенности людей с синдромом мозаичного Патау во много раз мягче, чем у людей с полным синдромом Патау.
Диагностика синдрома Патау
Младенцы с синдромом Патау могут иметь единственную пупочную артерию при рождении. Часто наблюдаются признаки врожденного порока сердца, такие как дефект межпредсердной перегородки, неправильное расположение сердца по направлению к груди, дефект межжелудочковой перегородки или открытый артериальный проток. Ультразвук или рентген желудочно-кишечного тракта могут показать вращение внутренних органов младенца.
КТ или МРТ головы младенца могут показать проблему со структурой его мозга. Проблема называется «голопроэнцефалия» и связана с соединением обеих сторон мозга младенца. Хромосомные исследования, в дополнение к этим тестам, могут помочь поставить окончательный диагноз синдрома Патау, а также дифференцировать синдром Патау, мозаицизм и частичный синдром Патау.
Лечение синдрома Патау
Лечение детей с синдромом Патау предполагает индивидуальное планирование.Формы лечения, которые получает конкретный человек, зависят от его конкретного состояния. Вмешательство хирургическим путем обычно воздерживается в течение первых нескольких месяцев жизни человека из-за высокой смертности детей с синдромом Патау. И родители, и медицинские работники должны взвесить решения, касающиеся чрезвычайных мер по продлению жизни, с серьезностью физических и неврологических дефектов, которые испытывает ребенок, и вероятностью послеоперационного выздоровления ребенка или его продолжительного выживания.
Синдром Патау включает в себя множество аномалий, несовместимых с жизнью. Более восьмидесяти процентов младенцев с синдромом не доживают до первого месяца жизни. Осложнения, связанные с синдромом, начинаются практически сразу, и многие младенцы страдают сердечными заболеваниями. Осложнения синдрома Патау могут включать проблемы с кормлением, затрудненное дыхание или недостаток дыхания, глухоту, сердечную недостаточность, судороги и проблемы со зрением.
Кто мы:
Disabled World — это независимое сообщество людей с ограниченными возможностями, созданное в 2004 году для предоставления новостей и информации об инвалидах людям с ограниченными возможностями, пожилым людям, их семьям и / или опекунам.