Синдром Шерешевского-Тернера: причины, симптомы и лечение
Синдром Шерешевского-Тернера – генетическая патология, которая обуславливается отсутствием в кариотипе индивида одной из хромосом половой системы. Характерным отличием болезни является то, что она поражает только девочек. Частота распространённости недуга составляет один случай на пять тысяч новорождённых девочек. Нередко при таком заболевании наблюдается самопроизвольное прерывание периода вынашивания ребёнка (на ранних сроках). По этой причине невозможно с точностью определить частоту, с которой встречается болезнь. Особенностью заболевания считается то, что оно имеет вполне благоприятный исход.
Признаками данного синдрома являются – низкорослость, различные пороки развития жизненно важных органов, косоглазие, наличие складок кожи на шее и деформация суставных сочленений. Это единственное генетическое заболевание, при котором представительницы женского пола живут с 45 хромосомами, вместо нормального кариотипа в 46 хромосом. Диагноз устанавливается благодаря специфическим клиническим особенностям. Кроме этого, возможно диагностировать такой недуг во время беременности.
Лечение синдрома комплексное и основывается на применении гормональной терапии с целью коррекции врождённых пороков. В международной классификации заболеваний (мкб 10) патология встречается под несколькими значениями – Q 96, непосредственно данное заболевание, Q 96.8 – другие вариации болезни.
Основная причина формирования синдрома Шерешевского-Тернера — это нарушение нормального кариотипа, при котором наблюдается отсутствие второй половой хромосомы. Механизм передачи заболевания до конца не выяснен. В медицинской сфере существует множество споров о его наследственности. Предрасполагающими факторами возникновения болезни являются:
- различные инфекции половой системы, перенесённые ранее будущей матерью;
- неблагоприятные условия окружающей среды, загазованность, загрязнённость;
- употребление женщиной в больших количествах спиртных напитков во время беременности;
- сильное излучение электромагнитного или ионизирующего характера;
- голодание или любое другое истощение организма, например, при тяжёлой болезни (зачастую в период перед зачатием).
В большинстве случаев появление синдрома наблюдается случайно — часто такие дети рождаются у абсолютно здоровых родителей. Это означает, что невозможно предугадать зачатие больного ребёнка или заранее провести профилактические мероприятия. Во время беременности можно узнать о болезни только при помощи анализа на кариотип.
Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера предусматривает наличие целого комплекса нарушений со стороны половой системы и строения жизненно важных органов. Исходя из характера генетической патологии, медицине известно несколько вариантов болезни:
- отсутствие второй половой хромосомы – такая форма недуга встречается чаще всего. Её особенностью является то, что в первом триместре яичники плода развиваются нормально. Но с увеличением срока беременности постепенно начинает проявляться замещение клеток будущей яйцеклетки соединительной тканью, а также начинают развиваться пороки внутренних органов. Помимо того, что такой тип является самым распространённым, он один из самых тяжёлых, симптомы ярко выражены, трудно поддаётся лечению, протекает с наличием осложнений;
- мозаичный – наиболее лёгкий тип недуга, поскольку тяжёлые пороки развития не наблюдаются, присутствуют лишь некоторые признаки специфической внешности, но их намного меньше, чем при предыдущей форме. Осложнения развиваются крайне редко. Лечению поддаётся хорошо;
- структурные изменения половых хромосом – диагностируется в редких случаях у женщин, имеющих нормальный кариотип, но одна их хромосом половой системы сильно повреждена. Характерные признаки болезни присутствуют, но их намного меньше, чем при первой форме.
Признаки синдрома Шерешевского-Тернера
У многих новорождённых девочек наблюдаются лишь незначительные симптомы, но у некоторых уже с рождения присутствует деформация кистей и стоп, а также складки кожи на задней поверхности шеи. К другим симптомам данного синдрома относят:
- специфическое выражение лица – мимика слаба, невозможность сморщить лоб, рот всегда полуоткрыт, нижняя губа немного отвисает;
- форма грудной клетки имеет вид бочки;
- отёчность нижних конечностей;
- низкий рост сочетается с высокими показателями массы тела;
- линия роста волос на шее расположена низко;
- соски локализуются значительно ниже и шире, нежели у здоровых детей;
- значительные нарушения полового созревания можно заметить в подростковом возрасте. Отсутствие месячных, волосяного покрова на лобке и в области подмышек, недоразвитость молочных желез;
- отставание в росте – чем старше становится ребёнок, тем ярче проявляется данный признак;
- искривление зубного ряда – наблюдается при всех генетических заболеваниях;
- косоглазие;
- задержка умственного развития – дети отличаются невнимательностью и плохой памятью;
- психические нарушения в виде эмоциональной нестабильности, депрессивных состояний, психозов, постоянная тревожность;
- пороки развития внутренних органов – сердечно-сосудистой системы, органов слуха, почек (зачастую наблюдается их удвоение).
Зачастую встречается такое последствие заболевания, как женское бесплодие. Но известны случаи, когда женщины, больные такой болезнью, могут забеременеть – традиционно или при помощи искусственного оплодотворения. Это возможно только у больных с мозаичным вариантом заболевания или в случае раннего гормонального лечения. В основном осложнения связаны с патологиями внутренних органов и систем:
- предрасположенность к раннему развитию заболеваний сердечно-сосудистой системы;
- инфекционно-воспалительные процессы в почках и мочевом пузыре;
- отит;
- возникновение злокачественных новообразований на кожном покрове;
- различные психологические проблемы;
- высокий риск развития сахарного диабета и ожирения.
При своевременной, правильной диагностике и комплексном лечении, люди с таким синдромом полностью адаптируются к жизни. Исключение составляют лица с тяжёлыми врождёнными пороками сердца и сосудов.
Несмотря на наличие специфических внешних признаков, сидром Шерешевского-Тернера диагностируется с некоторыми трудностями. Это обуславливается тем, что нередко встречается мозаичная форма недуга или структурные изменения двух половых хромосом. При таких вариантах вполне вероятно, что симптомы болезни могут отсутствовать. Подтвердить диагноз в таком случае может только специальный тест на кариотип – кариотипирование.
Основу диагностических мероприятий составляют:
- определение причин, по которым возник данный недуг;
- кариотипирование родителей и плода;
- УЗИ.
В первом случае необходимо провести детальный опрос родителей на предмет влияния вредных факторов или алкоголизма. Беседа с врачом-генетиком направлена на выяснение наличия генетических заболеваний, вплоть до четвёртого колена. Считается, что риск формирования синдрома велик, если кто-то в роду у одного из родителей страдал от хромосомных патологий. Если такая информация не подтверждается, то данный этап не имеет диагностической ценности.
Определение кариотипа родителей осуществляется не только при подозрениях у специалиста, но ещё может проводиться по желанию родителей. Процедура, по сути, напоминает обычный забор крови из вены для лабораторного изучения. Анализ проводится в период от 10 до 12 недели беременности. Процесс получения клеток плода для исследований проводится при помощи пункции, во время которой специальную иглу вводят в полость матки через переднюю брюшную стенку.
УЗИ плода во время беременности является незаменимым обследованием в диагностике многих внутриутробных заболеваний. Однозначно обнаружить такой синдром УЗИ не может, но оно предоставляет информацию о разнообразных признаках проявления недуга.
После рождения ребёнка с данным генетическим расстройством, необходимо провести дополнительные исследования:
- УЗИ внутренних органов;
- ЭКГ – для изучения работы сердца;
- ЭхоКГ – позволяет обнаружить аномалии сердечно-сосудистой системы;
- общий анализ крови – отражает множество процессов, протекающих во внутренних органах и системах;
- анализ урины – аналогично предыдущей процедуре отображает некоторые внутренние процессы;
- рентгенография кистей и костей стоп.
Помимо этого, больные с таким синдромом обязательно проходят осмотры у многих специалистов из разных сфер медицины. Таким образом, диагностика играет важную роль в определении кариотипа, формы заболевания и тактики лечения.
Как и многие хромосомные заболевания, данный синдром полностью вылечить невозможно. Основная задача терапии направлена на применение гормональных веществ и стимуляцию роста. В раннем детстве могут применяться лечебные массажи, курсы ЛФК и витаминные комплексы. В подростковом возрасте назначается заместительное лечение эстрогенами.
Помимо этого применяются:
- хирургическое вмешательство — для устранения пороков внутренних органов;
- пластические операции – ликвидация или уменьшение проявления внешних признаков болезни;
- психотерапевтическое лечение.
Специфической профилактики синдрома не существует. Родителям рекомендуется проводить анализ на кариотип перед зачатием и аналогичный тест для плода, при подозрениях специалиста.
Поделиться статьей:
Все ли корректно в статье с медицинской точки зрения?
Ответьте только в том случае, если у вас есть подтвержденные медицинские знанияпричины, симптомы и варианты лечения
Синдром Шерешевского-Тёрнера – генетическое заболевание, формирующееся на фоне отсутствия или дефекта X-хромосомы. Отклонение диагностируется в основном у девочек, у мальчиков описаны лишь единичные случаи. Возникновение патологии сопряжено с проявлениями гипогонадизма, отставанием в росте и развитии, поражениями опорно-двигательного аппарата и другими симптомами. Интенсивность клинических признаков определяется кариотипом пациента. При этом в большинстве случаев прогноз при заболевании благоприятный. Лечение позволяет скорректировать многие отклонения. Значимой проблемой при аномалии остается бесплодие, с трудом поддающееся терапии. В таких случаях пациенткам рекомендуется проведение ЭКО.
Причины возникновения синдрома Шерешевского-Тёрнера
Патология относится к числу генетических заболеваний. Она вызывается нарушением строения или количественной аномалией половой X-хромосомы. При этом исследователи отмечают различные вариации поражения. Данная проблема в большинстве случаев диагностируется у девочек, а у мальчиков регистрируется крайне редко, что связано с их низкой вынашиваемостью. Риски формирования недуга никак не ассоциированы с возрастом родителей или имеющимися у них патологиями. Течение и симптомы синдрома Шерешевского-Тёрнера определяются формой аномалии. Более чем у половины пациентов отмечается полное отсутствие второй Х-хромосомы. В 20% случаев изменения опосредованы структурными перестройками кариотипа плода. С той же инцидентностью встречается мозаичный тип проблемы, который сопряжен с минимальной интенсивностью клинических проявлений.

Точные причины возникновения генетического дефекта и развития синдрома Шерешевского-Тёрнера на сегодняшний день не установлены. Существуют предположения о негативном воздействии на плод во время беременности следующих факторов:
- Неблагоприятные экологические условия, например, загазованность воздуха, дефицит солнечного света или загрязнение окружающей среды химическими веществами.
- Ионизирующее излучение является одной из распространенных причин возникновения генетических мутаций.
- Сильные стрессы, а также физическое истощения организма матери на фоне интенсивных нагрузок и несбалансированного питания.
Несмотря на хромосомную природу заболевания, ему не свойственно наследование. Это связано с тем фактом, что большинство пациентов с патологией остаются бесплодными. Лишь малая часть людей с синдромом Шерешевского-Тёрнера способна иметь детей, если диагностирована мозаичная форма кариотипа нарушения.
Отличительные признаки заболевания
В ряде случаев поставить диагноз удается еще на этапе вынашивания малыша. С этой целью проводится пренатальный скрининг. Ребенок с данной патологией рождается с характерными особенностями, указывающими на генетический недуг:
- Небольшая масса и длина тела. Синдром Шерешевского-Тёрнера сопровождается формированием признаков недоношенности. Исключение составляют лишь пациенты с мозаичным вариантом патологии. При этом отставание в росте и весе становится все более заметным по мере развития ребенка. Эти показатели у больных значительно отличаются от массы и длины тела сверстников.
- Избыток кожи на шее, который образует крыловидные складки. Данный симптом считается специфическим для патологии. Дефект усугубляется также тем фактом, что этот отдел позвоночника у детей с заболеванием короче и шире, чем у здоровых.
- Лимфедема – отечность конечностей, возникающая вследствие нарушения нормального тока жидкости по сосудам. Поражение формируется преимущественно в области стоп младенца. Интенсивность симптома возрастает, когда ребенок начинает ходить, что связано с увеличением нагрузки на тазовые конечности.
- Деформации различных отделов скелета. У малышей с синдромом Шерешевского отмечается нарушение нормального строения локтевых суставов, изменение формы грудной клетки, которая приобретает бочкообразный вид, искривляются пальцы на руках.
- Общее беспокойство, снижение сосательного рефлекса и частые срыгивания лишь усугубляют клинические проявления проблемы.
- В ряде случаев патология сопряжена с другими внутриутробными дефектами. У пациентов с синдромом Шерешевского-Тёрнера зачастую диагностируются врожденные пороки сердца, аномалии строения почек и прочих внутренних органов.

По мере роста ребенка отмечается развитие и других симптомов. Клиническая картина синдрома основана на гипогонадизме, то есть недоразвитости половых желез. Подобный дефект провоцирует возникновение следующих признаков:
- Нарушения менструального цикла, вплоть до полной аменореи и отсутствия менархе. Подобная проблема связана с тем, что яичники у девочек представлены соединительнотканными тяжами и практически не имеют ооцитов. Половые железы не выполняют своих функций, поэтому происходят значительные гормональные нарушения, приводящие к бесплодию.
- Отставание в физическом развитии. Пациенты с синдромом Шерешевского-Тёрнера имеют невысокий рост, который, как правило, не превышает 150–155 см. Окружность черепа и грудной клетки также остается небольшой.
- Гормональные сбои обуславливают аномалии развития наружных половых органов и молочных желез. У девочек не происходит увеличения груди, как у здоровых подростков в пубертатный период. На фоне пониженной концентрации эстрогенов отмечается слабое развитие волосяного покрова в области лобка и подмышек. В ряде случаев растительность у пациенток развивается по мужскому типу, то есть на руках и в области верхней губы.
- Для недуга характерны и психологические симптомы. Отмечается запоздание в формировании личности ребенка. При этом интеллектуальные способности у пациентов с синдромом Тёрнера зачастую остаются на должном уровне. Девочки с данным заболеванием страдают от чрезмерной эмоциональной возбудимости и психологической неустойчивости, что обусловлено гипогонадизмом.
Необходимые диагностические исследования
Выявить заболевание представляется возможным еще до рождения ребенка, благодаря современным методам генетического анализа. Поэтому все обследования принято делить на пренатальные и постнатальные. К диагностике, используемой до рождения ребенка, относят:
- Кариотипирование будущих родителей, то есть анализ их ДНК. Наличие дефектов в материале матери или отца увеличивает риск развития патологии.
- Хотя при помощи УЗИ и невозможно поставить точный диагноз, оно широко применяется для выявления косвенных проявлений синдрома. Отечность плода, аномалии развития скелета, пороки сердца и другие признаки указывают на формирование генетического заболевания.
- Самым точным является проведение кариотипирования малыша. Для этого применяются различные инвазивные методики, позволяющие получить генетический материал ребенка. Данные процедуры сопряжены с риском инфицирования и прерывания беременности, поэтому решение об их проведении принимается совместно с гинекологом и генетиком.
После появления ребенка на свет поставить диагноз, как правило, не составляет труда. Это связано с тем, что в результате развития синдрома Шерешевского-Тёрнера у пациента формируются специфические симптомы, указывающие на проблему. Для подтверждения наличия заболевания проводятся следующие тесты:
- Анализы крови и мочи используются для определения гормонального статуса малыша, а также оценки работы внутренних органов.
- Для исключения наличия врожденных кардиальных поражений осуществляется УЗИ, в ходе которого делаются своеобразные фото сердца и его замеры, а также ЭКГ.

Методы лечения
Терапия заболевания зависит от кариотипа синдрома и варианта его клинического проявления. Поскольку основной проблемой при патологии остается бесплодие, широко применяются различные гормональные препараты. Для коррекции косметических дефектов, таких как крыловидные складки на шее, используются оперативные техники. У маленьких пациентов важную роль играет правильное кормление и обеспечение организма всеми необходимыми витаминами и нутриентами.
Гормонотерапия
Соматотропин широко используется у малышей с синдромом Шерешевского-Тёрнера. Назначение этого соединения позволяет стимулировать рост ребенка и набор массы тела. Для коррекции работы половой системы девочек применяется заместительная гормональная терапия препаратами эстрогенов. В ряде случаев эти средства используются вплоть до наступления у пациентки климакса. Вовремя начатое лечение синдрома Шерешевского-Тёрнера позволяет значительно снизить интенсивность его клинических проявлений.
Хирургическое вмешательство
Оперативные техники используются при различных проблемах. Они позволяют устранить косметические дефекты, такие как складки кожи в области шеи. Если формирование заболевания сопряжено с врожденными пороками сердца, осуществляется хирургическое вмешательство по протезированию клапанов и устранению дефектов перегородок.
Прочие методы
С целью профилактики прогрессирования патологических изменений и усугубления состояния маленьких пациентов используются неспецифические методы лечения, такие как специальные физические упражнения, массажные техники и физиотерапия. Важную роль играет правильное питание, а также использование витаминно-минеральных комплексов.
Бесплодие остается самым тревожным симптомом синдрома Тёрнера. У женщин возникают трудности с зачатием ребенка, поскольку отмечается ранняя овариальная недостаточность. Эта проблема связана со снижением количества или полным отсутствием фолликулов в яичниках, а также изменением работы половых желез и недостаточностью синтеза эстрогенов.
К сожалению, заместительная гормональная терапия не всегда дает положительные результаты. Эффективность такого лечения определяется причиной бесплодия. Если зачатие невозможно вследствие недостаточности выработки эстрогенов и высокой концентрации гонадотропинов, то использование медикаментозных средств приносит плоды. Однако в случаях, когда у женщины отсутствует яйцеклетка вследствие недоразвитости яичников и нехватки в них примордиальных фолликулов, гормональная терапия остается нерезультативной. В таких случаях существуют другие варианты зачатия:
- Использование донорской женской половой клетки.
- Оплодотворение in vitro с размещением зародыша в матке.
- Суррогатное материнство.
Важным этапом подготовки к беременности является проведение комплексной диагностики здоровья пациентки, направленного в первую очередь на исключение патологий сердца и почек.
Возможные последствия болезни и прогноз
Множество факторов влияет на течение и исход синдрома. Большое значение имеет кариотип патологии, поскольку он определяет наличие врожденных аномалий развития внутренних органов. Важную роль играет и своевременность лечения. В целом прогноз при синдроме Шерешевского-Тёрнера благоприятный, поскольку недуг существенно не влияет на продолжительность жизни. Исключение составляют лишь случаи, когда заболевание сочетается с пороками сердца и нарушением строения почек. Основной проблемой при недуге остается бесплодие. Лишь небольшая часть пациенток способна забеременеть. Однако во многих случаях женщинам с синдромом Шерешевского-Тёрнера успешно проводится экстракорпоральное оплодотворение (ЭКО), которое предполагает использование донорской яйцеклетки.
Отзывы
Алена, 26 лет, г. Москва
Я родилась с синдромом Шерешевского-Тёрнера. Диагноз врачи поставили еще в раннем детстве. Пришлось регулярно посещать медицинские учреждения, принимать гормональные таблетки. У меня мозаичный вариант болезни, поэтому признаки довольно сглаженные. Заместительную терапию препаратами эстрогена прохожу до сих пор.
Ольга, 34 года, г. Воронеж
Дочка появилась на свет недоношенной, с деформированной грудной клеткой и странными складками на шее. Врачи диагностировали синдром Шерешевского-Тёрнера. Это врожденная болезнь. Дочке назначили соматотропин, чтобы снизить интенсивность проявлений недуга. На продолжительность жизни патология не влияет. Будем продолжать лечение.
 Загрузка…
Загрузка…Синдром Тёрнера (Синдром Шерешевского — Тёрнера) [LifeBio.wiki]
Синдром Тернера, также известный как синдром Ульриха-Тернера, дизгенез гонад и 45,X, представляет собой состояние, при котором женщина частично или полностью утрачивает X-хромосому.1) Признаки и симптомы варьируются среди страдающих заболеванием. Часто они заключаются в короткой и перепончатой шее, низкой посадке ушей, низкой линии роста волос позади шеи, низком росте и раздутых руках и стопах при рождении. Обычно у страдающих синдромом отсутствуют периоды менструации, не развиваются молочные железы, они не способны иметь детей. Пороки сердца, сахарный диабет и низкий уровень тиреоидного гормона встречаются чаще. Большинство людей с синдромом Тернера имеет нормальные умственные способности. Многие, тем не менее, имеют проблемы с пространственным представлением, связанным с математикой. Часто возникают проблемы со слухом и зрением.2) Синдром Тернера обычно не наследуется от родителей.3) Отсутствуют известные факторы риска окружающей среды, возраст матери не играет роли. Синдром Тернера связан с хромосомной аберрацией, при которой вся или часть одной из X-хромосом утрачивается или меняется. В то время как большинство людей имеет 46 хромосом, люди с синдромом Тернера, как правило, имеют всего 45. Хромосомная аберрация может быть представлена только в некоторых клетках, в данном случае состояние носит название синдром Тернера с мозаицизмом. В данных случаях симптомов обычно меньше или, возможно, они вовсе отсутствуют. Диагностика основывается на физических признаках и генетическом тестировании.4) Лечения синдрома Тернера не существует. Тем не менее, лечение может облегчить симптомы. Инъекции человеческого гормона роста в детстве могут увеличить взрослый рост. Терапия замещения эстрогена может способствовать развитию молочных желез и таза. Часто требуется медицинский уход с целью управления другими проблемами со здоровьем, с которыми связывается синдром Тернера. Распространенность синдрома Тернера составляет от 1 на 2000 до 1 на 5000 женщин при рождении.5) Все регионы мира и культуры подвержены заболеванию практически в равной степени. Люди с синдромом Тернера имеют более короткую среднюю продолжительность жизни, что главным образом связано с проблемами с сердцем и сахарным диабетом. Генри Тернер первый описал состояние в 1938 г. В 1964 г. было выявлено, что синдром связан с хромосомной аберрацией.
Признаки и симптомы
Далее представлен перечень обычных симптомов синдрома Тернера. Важно заметить, что субъект может иметь любую комбинацию симптомов и маловероятно имеет все симптомы.
Низкий рост
Лимфедема (опухание) рук и стоп
Широкая грудь (щитовая грудь) и широко расставленные соски
Низкая линия роста волос
Низко посаженные уши
Бесплодие
Рудиментарные яичники в виде гонадного слоя (недоразвитые гонадальные структуры, которые позже становятся фиброзными)
Аменорея, или отсутствие периодов менструации
Повышенный вес, ожирение
Щитообразный торакс сердца
Укороченная пясть IV
Маленькие ногти на пальцах руки
Характерные лицевые особенности
Перепончатая шея в результате шейной гигромы в младенчестве
Стеноз аортального клапана
Коарктация аорты
Двустворчатый аортальный клапан
Подковообразная почка
Зрительные нарушения склеры, роговицы, глаукома и т.д.
Ушные инфекции и потеря слуха
Высокое соотношение талия-бедра (бедра не намного больше, чем талия)
Синдром дефицита внимания с гиперактивностью, или СДВГ (проблемы с концентрацией, памятью, вниманием, при этом гиперактивность наблюдается в основном у детей и подростков)
Нарушение невербальной обучаемости (проблемы с математикой, социальными навыками и пространственными соотношениями)
Другие особенности могут включать маленькую нижнюю челюсть (микрогнатия), вальгусную деформацию локтевого сустава,6) мягкие изогнутые ногти, ладонную складку и птоз верхнего века. Реже встречаются пигментные невусы, потеря слуха и высокий подъем неба (узкая верхняя челюсть). Синдром Тернера сам по себе проявляется по-разному у каждой женщины, подверженной заболеванию. В то время как большая часть нарушений безвредна, с синдромом могут быть связаны значительные медицинские проблемы.
Пренатальные
Несмотря на очень хороший постнатальный прогноз, 99% случаев зачатий с синдромом Тернера предположительно заканчиваются выкидышем или мертворождением, при этом до 15% всех выкидышей имеет кариотип 45,X. Среди случаев, которые обнаруживаются регулярным амниоцентезом или биопсией ворсин хориона, одно исследование выявило, что распространенность синдрома Тернера среди протестированных случаев беременности было в 5,58 и 13,3 раз выше, чем среди здоровых младенцев той же группы людей.7)
Сердечно-сосудистые
Распространенность аномалий сердечно-сосудистой системы
Распространенность аномалий сердечно-сосудистой системы среди пациентов с синдромом Тернера находится в диапазоне от 17% (Ландин-Вильгельмсен и др., 2001) до 45% (Доусон-Фолк и др., 1992). Изменчивость, обнаруженная в различных исследованиях, главным образом объясняется вариациями неинвазивных методов, применяемых для анализа, и типами повреждений, которые они характеризуют (Го и др., 2004). Тем не менее, Сиберт, 1998 г., свидетельствует, что это может объясняться всего лишь небольшим числом субъектов в большинстве исследований. Различные кариотипы могут давать различную распространенность аномалий сердечно-сосудистой системы. Два исследования обнаружили распространенность аномалий сердечно-сосудистой системы в 30% и 38%8) в группе чистой 45,X моносомии. Но, учитывая кариотипы других групп, они сообщили о распространенности в 24,3% и 11% у пациентов с мозаичной X моносомией и распространенности в 11% у пациентов с X хромосомными аномалиями строения. Более высокая распространенность в группе чистой 45,X моносомии в первую очередь связана со значительной разницей в распространенности аномалий аортального клапана и коарктации аорты, двух наиболее частых аномалий сердечно-сосудистой системы.
Врожденный порок сердца
Наиболее часто наблюдаются врожденные обструктивные повреждения левой части сердца, что ведет к снижению кровотока в этой части сердца. Они охватывают двустворчатый аортальный клапан и коарктацию (сужение) аорты. Сиберт, 1998 г., обнаружила, что более 50% аномалий сердечно-сосудистой системы, наблюдаемых в ее исследовании субъектов с синдромом Тернера, составляли двустворчатые аортальные клапаны и коарктация аорты, в отдельности или в сочетании. Другие врожденные аномалии сердечно-сосудистой системы, такие как частичный аномальный венозный отток и стеноз аортального клапана или регургитация аорты, чаще встречаются у страдающих синдромом Тернера, чем у общей совокупности населения. Гипоплазия левой половины сердца представляет собой наиболее серьезное сокращение левосторонних структур.
Двустворчатый аортальный клапан
До 15% взрослых с синдромом Тернера имеют двустворчатые аортальные клапаны, означающие, что имеется только две, вместо трех, части клапанов основного кровеносного сосуда, идущего от сердца. Поскольку двустворчатые клапаны способны надлежащим образом регулировать кровоток, данное состояние может быть не обнаружено в отсутствие регулярного обследования. Тем не менее, двустворчатые клапаны более вероятно изнашиваются и в дальнейшем перестают функционировать. Также возникает кальцификация клапанов, которая может привести к прогрессирующей клапанной дисфункции, что вызывает стеноз аорты и регургитацию.9) С распространенностью от 12,5% до 17,5% (Доусон-Фолк и др., 1992), двустворчатый аортальный клапан представляет собой наиболее часто наблюдаемую врожденную аномалию, влияющую на сердце при данном синдроме. Обычно возникает в отдельности, но может наблюдаться в сочетании с другими аномалиями, в частности, коарктацией аорты.
Коарктация аорты
От 5% до 10% рожденных с синдромом Тернера имеют коарктацию аорты, врожденное сужение нисходящей части аорты, обычно удаленной от левой подключичной артерии (артерия, которая ответвляется от дуги аорты к левой руке) и расположенной напротив протока (также называется «юкстадактальная»). Расчетные показатели распространенности данной аномалии среди пациентов с синдромом Тернера находятся в диапазоне от 6,9% до 12,5% . Коарктация аорты у женщин свидетельствует о синдроме Тернера и о потребности в дальнейших анализах, таких как кариотип.
Частичный аномальный венозный отток
Данная аномалия представляет собой сравнительно редкое врожденное заболевание сердца по отношению к общей совокупности населения. Распространенность данной аномалии также является низкой (примерно 2,9%) при синдроме Тернера. Тем не менее, сравнительный риск составляет 320 по сравнению с общей совокупностью населения. Что странно, синдром Тернера связывается с необычными формами частичного аномального венозного оттока.10) Управление заболеванием у пациентов с синдромом Тернера жизненно необходимо, учитывая, что данная побочная аномалия сердечно-сосудистой системы при синдроме Тернера приводит к повышенной предрасположенности к бактериальному эндокардиту. Следовательно, когда выполняются процедуры с высоким риском развития эндокардита, такие как профессиональная чистка зубов, для профилактики должны приниматься антибиотики. Синдром Тернера часто связывается с устойчивой гипертензией, в некоторых случаях в детстве. У большинства пациентов с синдромом Тернера и гипертензией отсутствует специфическая причина. В остальном она обычно связана с сердечно-сосудистыми или почечными аномалиями, включая коарктацию аорты.
Дилатация, расслоение и разрыв аорты
Два исследования свидетельствуют о дилатации аорты при синдроме Тернера, обычно охватывающей основание восходящей части дуги аорты и изредка распространяющейся через дугу на нисходящую часть аорты, либо на место предыдущей коарктации зажившей аорты.11)
Аллен и др., 1986 г., которые изучали 28 девочек с синдромом Тернера, обнаружили значительно больший средний диаметр основания аорты у пациентов с синдромом Тернера, чем у контрольной группы (оценивался по отношению к площади поверхности тела). Тем не менее, диаметр основания аорты, обнаруженный у пациентов с синдромом Тернера, все же находился в диапазоне предельных значений.
Это было подтверждено исследованием Доусона-Фолка и др., 1992 г., которые изучали 40 пациентов с синдромом Тернера. Они представили в основном аналогичные результаты: больший средний диаметр основания аорты, который все же остается в пределах нормального диапазона в отношении площади поверхности тела.
Сиберт, 1998 г., обращает внимание на недоказанность того, что диаметр основания аорты, который сравнительно велик в отношении площади поверхности тела, но все же остается в пределах нормального диапазона, предполагает риск прогрессирующей дилатации.12)
Распространенность аортальных аномалий
Распространенность дилатации основания аорты у пациентов с синдромом Тернера находится в диапазоне от 8,8% до 42%. Даже если не каждая дилатация основания аорты обязательно прогрессирует до расслоения аорты (кольцевой или поперечный разрыв интимы), могут возникнуть осложнения, такие как расслоение и разрыв аорты, которые приводят к смерти. Течение развития дилатации основания аорты до сих пор не известно, но известен факт, что это связано с расслоением и разрывом аорты, которые имеют высокую долю смертности. Расслоению аорты подвержены от 1% до 2% пациентов с синдромом Тернера. Как результат, дилатация основания аорты должна быть серьезно принята во внимание, поскольку она может привести к фатальному расслоению аорты. Настоятельно рекомендуется регулярное обследование.
Факторы риска в отношении разрыва аорты
Хорошо известно, что аномалии сердечно-сосудистой системы (обычно двустворчатый аортальный клапан, коарктация аорты и некоторые другие побочные сердечно-сосудистые аномалии) и гипертензия предрасполагают к дилатации и расслоению аорты у общей совокупности населения. В то же время выявлено, что они представляют факторы риска при синдроме Тернера. Более того, аналогичные факторы риска были обнаружены у более чем 90% пациентов с синдромом Тернера, у которых развилась дилатация аорты. Только небольшое число пациентов (примерно 10%) не имеют явно предрасполагающих факторов риска. Важно отметить, что риск гипертензии в 3 крат выше у пациентов с синдромом Тернера. По причине связи с расслоением аорты, кровяное давление должно постоянно контролироваться, а гипертензия должна подвергаться агрессивному лечению с целью поддержания кровяного давления ниже 140/80 мм рт.ст. Было выявлено, что, как и в случае других аномалий сердечно-сосудистой системы, осложнения дилатации аорты в достаточной степени связаны с кариотипом 45,X.
Патогенез расслоения и разрыва аорты
Точная роль, которую данные факторы риска играют в процессе, ведущем к таким фатальным осложнениям, до сих пор достаточно не ясна. Дилатация основания аорты предположительно связана с мезенхимальным дефектом как патологическим свидетельством кистозного медионекроза аорты, обнаруженным несколькими исследованиями. Связь между аналогичным дефектом и дилатацией аорты точно установлена при таком состоянии как синдром Марфана. Кроме того, аномалии в других мезенхимальных тканях (костный матрикс и лимфатические сосуды) свидетельствуют об аналогичном исходном мезенхимальном дефекте у пациентов с синдромом Тернера. Тем не менее, отсутствуют доказательства, свидетельствующие, что пациенты с синдромом Тернера имеют в значительной степени более высокий риск дилатации и расслоения аорты в отсутствие предрасполагающих факторов. Таким образом, риск расслоения аорты при синдроме Тернера является скорее результатом аномалий сердечно-сосудистой системы и гемодинамических факторов риска, чем отражением наследственной аномалии соединительной ткани (Сиберт, 1998). Tечение развития дилатации основания аорты не известно, но поскольку она имеет летальный потенциал, данная аортальная аномалия должна тщательно обследоваться.
Скелетные
Нормальное скелетное развитие ингибируется в связи с широким множеством факторов, главным образом, гормональных. Средний рост женщины с синдромом Тернера, в отсутствие лечения гормоном роста, составляет 4 фута 7 дюймов (140 см). Пациенты с мозаицизмом Тернера могут достигать нормальный средний рост. Четвертая метакарпальная кость (четвертый палец ноги и безымянный палец) может быть необычно короткой, как и пятая. В связи с нарушенной выработкой эстрогена, у многих страдающих синдромом Тернера развивается остеопороз. Это в дальнейшем может уменьшать рост, а также ухудшить искривление позвоночника, что потенциально ведет к сколиозу. Он также связывается с повышенным риском переломов костей.
Почки
Приблизительно одна треть всех женщин с синдромом Тернера имеет одну из трех аномалий почек:
Единственная подковообразная почка с одной стороны организма.
Атипичная система сбора мочи.
Плохой кровоток, идущий к почкам.
Некоторые из данных состояний могут быть исправлены хирургически. Даже при наличии данных аномалий почки большинства женщин с синдромом Тернера могут функционировать нормально. Тем не менее, как отмечалось выше, проблемы с почками могут быть связаны с гипертензией.
Щитовидная железа
Приблизительно одна треть всех женщин с синдромом Тернера имеет заболевание щитовидной железы. Обычно оно представлено гипотиреозом, а именно тиреоидитом Хашимото. Когда обнаружен, он может легко поддаваться лечению добавками тиреоидного гормона.
Сахарный диабет
Женщины с синдромом Тернера имеют незначительно повышенный риск развития сахарного диабета типа 1 в детстве и в значительной степени повышенный риск развития сахарного диабета типа 2 во взрослом возрасте. Риск развития сахарного диабета типа 2 может быть существенно снижен за счет поддержания здорового веса.
Когнитивная функция
Синдром Тернера обычно не вызывает умственной отсталости или нарушения когнитивной функции. Тем не менее, сложности обучения распространены среди женщин с синдромом Тернера, в частности, специфическая сложность понимания пространственных связей, а также нарушение способности к невербальному обучению. Также может он проявляться как сложность с двигательным контролем или математикой. В то время как не поддается лечению, в большинстве случаев это не вызывает трудностей в повседневной жизни. Большинство пациентов с синдромом Тернера нанимаются на работу как полноценные люди и ведут продуктивную жизнь. Также существует редкая разновидность синдрома Тернера, известная как «Кольцевой-X синдром Тернера», которые имеет примерно 60-процентную связь с умственной отсталостью. Данная разновидность охватывает приблизительно 2–4% всех случаев синдрома Тернера.13)
Репродуктивная система
Женщины с синдромом Тернера практически в всех случаях бесплодны. В то время как некоторые женщины с синдромом Тернера могут успешно зачать ребенка и перенести беременность, это достаточно редко и, как правило, ограничивается теми женщинами, чей кариотип не соответствует 45,X. Даже когда такая беременность возникает, существует превышающий средний риск выкидыша или врожденных дефектов, включая синдромы Тернера или Дауна. Некоторые женщины с синдромом Тернера, неспособные зачать без медицинского вмешательства, могут использовать экстракорпоральное оплодотворение или другие средства преодоления бесплодия.14) Терапия замещения эстрогена, как правило, применяется с целью стимулировать рост вторичных половых признаков тогда, когда должно начаться половое созревание. В то время как достаточно небольшое число женщин с синдромом Тернера менструируют самопроизвольно, эстрогеновая терапия требует регулярного выведения слизистой оболочки матки («кровотечение отмены») с целью предотвращения ее чрезмерного роста. Кровотечение отмены может вызываться ежемесячно, как менструация, или менее часто, обычно каждые три месяца, по желанию пациента. Эстрогеновая терапия не делает женщину с нефункциональными яичниками способной к деторождению, но играет важную роль в искусственном оплодотворении; здоровье матки необходимо поддерживать с помощью эстрогена, если пригодная для оплодотворения женщина с синдромом Тернера желает использовать экстракорпоральное оплодотворение (с использованием донорских ооцитов). Синдром Тернера представляет собой причину первичной аменореи, синдрома истощения яичников (гипергонадотропный гипогонадизм), полосковидных гонад и бесплодия. Невозможность развития вторичных половых признаков (сексуальный инфантилизм) является типичной для состояния. В особенности в мозаичных случаях синдрома Тернера, который затрагивает Y-хромосому (например, 45,X/46,XY) и связан с риском развития злокачественной опухоли яичника (наиболее распространена гонадобластома), рекомендуется гонадэктомия. Синдром Тернера характеризуется первичной аменореей, синдромом истощения яичников, полосковидными гонадами и бесплодием. Тем не менее, технологии (особенно донорство ооцитов) обеспечивают данным пациентам возможность зачать ребенка. Поскольку все больше женщин с синдромом Тернера доводит беременность до завершения благодаря современным техникам лечения бесплодия, было отмечено, что беременность может нести риск сердечно-сосудистых осложнений для матери. Более того, несколько исследований свидетельствует о повышенном риске расслоения аорты при беременности. Сообщалось даже о трех случаях летального исхода. Влияние эстрогена подвергалось исследованиям, но остается неясным. Предполагается, что высокий риск расслоения аорты во время беременности у женщин с синдромом Тернера может быть связан скорее с повышенной гемодинамической нагрузкой, чем с высоким уровнем эстрогена. Безусловно, данные результаты важны и должны приниматься во внимание в отношении беременных пациентов с синдромом Тернера.
Причина
Синдром Тернера представляет собой последствие отсутствия двух полных копий X-хромосомы в некоторых или во всех клетках. Атипичные клетки могут иметь только одну хромосому X (моносомия) (45,X) или могут быть подвержены одному из нескольких типов частичной моносомии, как делеция короткой p дуги одной X-хромосомы (46,X,дел(Xp) или наличие изохромосомы с двумя q дугами (46,X,i(Xq). У мозаичных субъектов клетки с X-моносомией (45,X) могут наблюдаться наряду с нормальными клетками (46,XX), клетками, которые имеют частичную моносомию, или клетками, которые имеют Y-хромосому (46,XY). Наличие мозаицизма, по расчетам, сравнительно часто встречается у подверженных синдрому Тернера пациентов (67–90%).15)
Наследование
В большинстве случаев, когда возникает моносомия, X-хромосома берется от матери. Это может быть связано с нерасхождением у отца. Также у отцов часто обнаруживаются мейотические ошибки, которые ведут к образованию хромосомы X с делецией p дуги и патологической Y-хромосомы. Изохромосома X или кольцевая хромосома X, с другой стороны, формируются с равной встречаемостью у обоих родителей. В целом, функциональная X-хромосома в большинстве случаев исходит от матери. В большинстве случаев синдром Тернера представляет собой случайное событие, и для родителей ребенка с синдромом Тернера риск повторения для последующих беременностей не повышается. Редкие исключения могут включать наличие сбалансированной транслокации X-хромосомы у родителя, либо случаи, в которых мать имеет 45,X мозаицизм, ограниченный ее первичными половыми клетками.16)
Диагностика
Пренатальная
Синдром Тернера может быть диагностирован во время беременности с помощью амниоцентеза или биопсии ворсин хориона. Как правило, плод с синдромом Тернера может быть выявлен посредством отклоняющихся от нормы результатов ультразвукового исследования (например, порок сердца, аномалия почек, шейная гигрома, асцит). В исследовании 19 европейских журналов учета, 67,2% пренатально диагностированных случаев синдрома Тернера были обнаружены за счет аномалий при ультразвуковом исследовании. В 69,1% случаев была представлена одна аномалия, а в 30,9% случаев – две и более аномалии. Повышенный риск развития синдрома Тернера также может быть выявлен с помощью отклоняющихся от нормы триады или квадруплета в обследовании материнской крови. Плод, диагностированный посредством положительного анализа материнской сыворотки, чаще имеет мозаичный кариотип, чем тот, диагноз которого основан на аномалиях ультразвукового исследования, и, наоборот, субъекты с мозаичными кариотипами менее вероятно имеют связанные ультразвуковые аномалии. Хотя риск рецидива не повышается, семьям, в которых имеется случай беременности или ребенка с синдромом Тернера, часто рекомендуется генетическое консультирование.
Постнатальная
Синдром Тернера может быть диагностирован постнатально в любом возрасте. Часто он диагностируется при рождении в связи с проблемами с сердцем, необычно широкой шеей и отеком рук и стоп. Тем не менее, часто встречается, что он остается недиагностированным в течение нескольких лет, обычно пока девочка не достигнет возраста полового созревания/пубертатного возраста и не сможет начать развиваться надлежащим образом (изменения, связанные с половым созреванием, не произойдут). В детстве о синдроме Тернера может свидетельствовать низкий рост. Анализ, носящий название кариотип или анализ хромосом, анализирует хромосомный состав субъекта. Это предпочтительный анализ с целью диагностики синдрома Тернера.
Лечение
Поскольку синдром Тернера представляет собой хромосомное состояние, лечения не существует. Тем не менее, для облегчения симптомов может быть предпринято множество мер. Например:17)
Врачи могут использовать инъекцию гормона роста, известного как Генотропин («Pfizer»)
- Гормон роста, в отдельности или с низкой дозой андрогена, увеличит рост и, возможно, приведет к достижению взрослого роста. Гормон роста утвержден Управлением по контролю за продуктами питания и лекарственными средствами США для лечения синдрома Тернера и оплачивается многими программами страхования. Существуют доказательства, что он эффективен даже у только начавших ходить детей.18)
Терапия замещения эстрогена, такая как пероральные противозачаточные средства, применялась с тех пор, когда состояние было описано в1938 г., с целью содействия развитию вторичных половых признаков. Эстрогены крайне важны для обеспечения целостности костей, здоровья сердечно-сосудистой системы и тканей. Женщины с синдромом Тернера, не демонстрирующие спонтанного полового созревания и не проходящие лечение с помощью эстрогена, находятся в группе повышенного риска развития остеопороза и сердечных заболеваний.
Современные репродуктивные технологии также применяются с целью помочь женщинам с синдромом Тернера в их желании зачать ребенка. Например, для образования эмбриона может использоваться донорская яйцеклетка, при этом ребенок будет вынашиваться женщиной с синдромом Тернера.
Маточная зрелость положительно связана с годами применения эстрогена, историей спонтанного менархе и отрицательно связана с отсутствием текущей терапии замещения гормона.
Эпидемиология
Примерно в 99 процентов случаев зачатия при синдроме Тернера наблюдается самопроизвольное прерывание беременности во время первого триместра.19) Синдром Тернера охватывает приблизительно 10 процентов от общего числа выкидышей в США. Встречаемость синдрома Тернера у рождающихся здоровых девочек предположительно составляет 1 на 2000.
История
Синдром получил название в честь Генри Тернера, эндокринолога из Иллинойса, который описал состояние в 1938 г.20) В Европе состояние часто называют синдромом Ульриха-Тернера или даже синдром Бонневи-Ульриха-Тернера, признавая, что ранние случаи синдрома также были описаны европейскими врачами. Первое опубликованное сообщение о женщине с кариотипом 45,X было сделано в 1959 г. доктором Чарльзом Фордом и его коллегами в Харвелле, Оксфордшир и госпитале Гая в Лондоне.[44] Он был обнаружен у 14-летней девочки с признаками синдрома Тернера.
:Tags
Читать еще: Бендамустин (Рибомустин) , Каркаде (Суданская роза) , Наттокиназа , Никотин , Остролодочник серповидный ,
Список использованной литературы:
1) «Turner Syndrome: Overview». Eunice Kennedy Shriver National Institute of Child Health and Human Development. 3 April 2013. Retrieved 15 March 2015. 2) Sybert VP, McCauley E; McCauley (September 2004). «Turner’s syndrome». N. Engl. J. Med. 351 (12): 1227–38. doi:10.1056/NEJMra030360. PMID 15371580. 3) «How many people are affected or at risk?». Eunice Kennedy Shriver National Institute of Child Health and Human Development. 30 November 2012. Retrieved 15 March 2015. 4) «How do health care providers diagnose Turner syndrome?». Eunice Kennedy Shriver National Institute of Child Health and Human Development. 30 November 2012. Retrieved 15 March 2015. 5) Marino, Bradley S. (2013). Blueprints pediatrics (Sixth edition. ed.). Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins. p. 319. ISBN 9781451116045. 6) Chapter on Amenorrhea in: Bradshaw, Karen D.; Schorge, John O.; Schaffer, Joseph; Lisa M. Halvorson; Hoffman, Barbara G. (2008). Williams’ Gynecology. McGraw-Hill Professional. ISBN 0-07-147257-6. 7) Gravholt CH, Juul S, Naeraa RW, Hansen J; Juul; Naeraa; Hansen (1996-01-06). «Prenatal and postnatal prevalence of Turner’s syndrome: a registry study». BMJ (Clinical research ed.) 312 (7022): 16–21. doi:10.1136/bmj.312.7022.16. PMC 2349728. PMID 8555850. 8) Gøtzsche CO, Krag-Olsen B, Nielsen J, Sørensen KE, Kristensen BO; Krag-Olsen; Nielsen; Sørensen; Kristensen (Nov 1994). «Prevalence of cardiovascular malformations and association with karyotypes in Turner’s syndrome». Arch Dis Child. 71 (5): 433–6. doi:10.1136/adc.71.5.433. PMC 1030059. PMID 7826114. 9) Elsheikh M, Dunger DB, Conway GS, Wass JA; Dunger; Conway; Wass (Feb 2002). «Turner’s syndrome in adulthood». Endocr. Rev. 23 (1): 120–40. doi:10.1210/er.23.1.120. PMID 11844747. 10) Prandstraller D, Mazzanti L, Picchio FM, Magnani C, Bergamaschi R, Perri A, Tsingos E, Cacciari E; Mazzanti; Picchio; Magnani; Bergamaschi; Perri; Tsingos; Cacciari (1999). «Turner’s syndrome: cardiologic profile according to the different chromosomal patterns and long-term clinical follow-Up of 136 nonpreselected patients». Pediatr Cardiol 20 (2): 108–12. doi:10.1007/s002469900416. PMID 9986886. 11) Lin AE, Lippe B, Rosenfeld RG; Lippe; Rosenfeld (Jul 1998). «Further delineation of aortic dilation, dissection, and rupture in patients with Turner syndrome». Pediatrics 102 (1): e12. doi:10.1542/peds.102.1.e12. PMID 9651464. 12) Sybert VP (Jan 1998). «Cardiovascular malformations and complications in Turner syndrome». Pediatrics 101 (1): E11. doi:10.1542/peds.101.1.e11. PMID 9417175. 13) Berkovitz G, Stamberg J, Plotnick LP, Lanes R; Stamberg; Plotnick; Lanes (Jun 1983). «Turner syndrome patients with a ring X chromosome». Clin Genet. 23 (6): 447–53. doi:10.1111/j.1399-0004.1983.tb01980.x. PMID 6883789. 14) Hovatta O (1999). «Pregnancies in women with Turner’s syndrome». Annals of Medicine 31 (2): 106–10. doi:10.3109/07853899908998785. PMID 10344582. 15) Crespi B (2008). «Turner syndrome and the evolution of human sexual dimorphism». Evolutionary Applications 1 (3): 449–461. doi:10.1111/j.1752-4571.2008.00017.x. 16) Frías JL, Davenport ML; Davenport; Committee on Genetics Section on Endocrinology (2003). «Health Supervision for Children with Turner Syndrome». Pediatrics 111 (3): 692–702. doi:10.1542/peds.111.3.692. PMID 12612263. 17) Turner Syndrome Society of the United States. «FAQ 6. What can be done?». Retrieved 2007-05-11. 18) Davenport ML, Crowe BJ, Travers SH, Rubin K, Ross JL, Fechner PY, Gunther DF, Liu C, Geffner ME, Thrailkill K, Huseman C, Zagar AJ, Quigley CA (2007). «Growth hormone treatment of early growth failure in toddlers with Turner syndrome: a randomized, controlled, multicenter trial». J Clin Endocrinol Metab. 92 (9): 3406–16. doi:10.1210/jc.2006-2874. PMID 17595258. 19) Urbach A, Benvenisty N; Benvenisty (2009). «Studying early lethality of 45,XO (Turner’s syndrome) embryos using human embryonic stem cells». PLoS ONE 4 (1): e4175. doi:10.1371/journal.pone.0004175. PMC 2613558. PMID 19137066. 20) Turner HH (1938). «A syndrome of infantilism, congenital webbed neck, and cubitus valgus». Endocrinology 23 (5): 566–74. doi:10.1210/endo-23-5-566.
синдром_тернера.txt · Последние изменения: 2019/08/06 14:21 — nataly
Синдром Шерешевского-Тернера — причины, симптомы, лечение
 Внутриутробное развитие ребенка – сложный и достаточно длительный процесс. Далеко не всегда он завершается рождением абсолютно здорового малыша. Среди возможных нарушений развития отдельной строкой стоят хромосомные аномалии. Они различаются по тяжести поражения плода и могут вызывать у него как физические, так, впоследствии, и психические отклонения. Одной из подобных аномалий является синдром Шерешевского-Тернера, который развивается преимущественно у девочек.
Внутриутробное развитие ребенка – сложный и достаточно длительный процесс. Далеко не всегда он завершается рождением абсолютно здорового малыша. Среди возможных нарушений развития отдельной строкой стоят хромосомные аномалии. Они различаются по тяжести поражения плода и могут вызывать у него как физические, так, впоследствии, и психические отклонения. Одной из подобных аномалий является синдром Шерешевского-Тернера, который развивается преимущественно у девочек.
Синдром Шерешевского-Тернера, причины
Болезнь, известная как синдром Шерешевского-Тернера, является не слишком распространенной патологией. Данная хромосомная аномалия была описана еще в начале прошлого века. За прошедшие годы болезнь достаточно изучена различными специалистами. Свое название она получила по именам двух ученых, выявивших основные особенности этой патологии.
Синдром Шерешевского-Тернера, причины которого были раскрыты позже, только в середине прошлого века, обусловлен неполноценностью хромосомного набора, полученного ребенком от родителей. Кариотип человека состоит из 23 пар хромосом, последняя из которых определяет половую принадлежность. Она может быть представлена либо двумя одинаковыми – XX, либо двумя разными – XY-хромосомами. В первом случае организм будет развиваться по женскому типу, во втором – по мужскому.
Повреждение или же полное отсутствие в паре одной X-хромосомы и обуславливает развитие у ребенка синдрома Шерешевского-Тернера, признаки которого проявляются уже с рождения. При этом в начале внутриутробного развития хромосомный набор имеет нормальный состав. Однако половые клетки быстро регрессируют и, в результате, младенец появляется на свет, имея неполноценный кариотип.
Синдром Шерешевского-Тернера, симптомы
Уже в период новорожденности у ребенка можно увидеть характерные признаки отклонения в физическом развитии. Он отличается малым ростом и низкой массой тела. На боковой поверхности шеи имеются крыловидные кожные складки, граница роста волос значительно снижена, ушные раковины недоразвиты, отмечается отечность стоп. Отличительной особенностью такого ребенка, по мере его взросления, становится очень низкий рост.
При синдроме Шерешевского-Тернера симптомы достаточно многочисленны. Наиболее характерными являются три из них. Это половое недоразвитие, кожные складки на шее и аномалия локтевых суставов. Также могут наблюдаться глухота, врожденный птоз – опущение века, пороки сердца. Хотя интеллект у большинства больных с данной аномалией сохранен, все же частота умственного недоразвития среди них значительно выше, чем среди обычных детей.
По мере взросления больного с синдромом Шерешевского-Тернера симптомы отклонения в половом развитии проявляются все ярче. Отсутствуют вторичные половые признаки, отмечается аменорея вследствие недоразвития матки и яичников.
Синдром Шерешевского-Тернера, лечение
 Как хромосомная аномалия, синдром Шерешевского-Тернера лечению подлежит только симптоматическому. Чтобы больная могла вести нормальный образ жизни, ей все же необходимо получать медицинскую помощь. В раннем возрасте лечение направлено на стимуляцию роста, при этом назначаются анаболические стероиды и соматотропин. У ребенка оперативным путем производят удаление крыловидных кожных складок на шее. Необходимую помощь оказывают при врожденных пороках развития, представляющих опасность для жизни, в первую очередь – при сердечных аномалиях.
Как хромосомная аномалия, синдром Шерешевского-Тернера лечению подлежит только симптоматическому. Чтобы больная могла вести нормальный образ жизни, ей все же необходимо получать медицинскую помощь. В раннем возрасте лечение направлено на стимуляцию роста, при этом назначаются анаболические стероиды и соматотропин. У ребенка оперативным путем производят удаление крыловидных кожных складок на шее. Необходимую помощь оказывают при врожденных пороках развития, представляющих опасность для жизни, в первую очередь – при сердечных аномалиях.
Во время наступления периода полового созревания у девочки с синдромом Шерешевского-Тернера лечение осуществляется с помощью женских гормонов. Это способствует появлению вторичных половых признаков, формированию телосложения по женскому типу, некоторому росту матки и яичников. Проведение данной терапии в сочетании с современными репродуктивными технологиями позволяет впоследствии благополучно выносить ребенка.
Таким образом, синдром Шерешевского-Тернера, признаки которого явственно говорят о бесплодии, на сегодняшний день не является приговором для женщины. Она может иметь достаточно полноценную жизнь, несмотря на сложное наследственное заболевание.
Конечно, рождение ребенка с синдромом Шерешевского-Тернера, как и с любой другой аномалией, является большим потрясением для родителей. Однако своевременная и систематическая медицинская помощь позволяет значительно компенсировать недостатки в его развитии. В этой ситуации, помимо лечебной составляющей, для подрастающего ребенка очень важна поддержка близких ему людей.
Видео с YouTube по теме статьи:
Информация является обобщенной и предоставляется в ознакомительных целях. При первых признаках болезни обратитесь к врачу. Самолечение опасно для здоровья!
Знаете ли вы, что:Во время чихания наш организм полностью прекращает работать. Даже сердце останавливается.
Первый вибратор изобрели в 19 веке. Работал он на паровом двигателе и предназначался для лечения женской истерии.
Кариес – это самое распространенное инфекционное заболевание в мире, соперничать с которым не может даже грипп.
Самая высокая температура тела была зафиксирована у Уилли Джонса (США), который поступил в больницу с температурой 46,5°C.
Стоматологи появились относительно недавно. Еще в 19 веке вырывать больные зубы входило в обязанности обычного парикмахера.
В нашем кишечнике рождаются, живут и умирают миллионы бактерий. Их можно увидеть только при сильном увеличении, но, если бы они собрались вместе, то поместились бы в обычной кофейной чашке.
Желудок человека неплохо справляется с посторонними предметами и без врачебного вмешательства. Известно, что желудочный сок способен растворять даже монеты.
У 5% пациентов антидепрессант Кломипрамин вызывает оргазм.
Если улыбаться всего два раза в день – можно понизить кровяное давление и снизить риск возникновения инфарктов и инсультов.
Препарат от кашля «Терпинкод» является одним из лидеров продаж, совсем не из-за своих лечебных свойств.
В Великобритании есть закон, согласно которому хирург может отказаться делать пациенту операцию, если он курит или имеет избыточный вес. Человек должен отказаться от вредных привычек, и тогда, возможно, ему не потребуется оперативное вмешательство.
Когда влюбленные целуются, каждый из них теряет 6,4 ккалорий в минуту, но при этом они обмениваются почти 300 видами различных бактерий.
Общеизвестный препарат «Виагра» изначально разрабатывался для лечения артериальной гипертонии.
Существуют очень любопытные медицинские синдромы, например, навязчивое заглатывание предметов. В желудке одной пациентки, страдающей от этой мании, было обнаружено 2500 инородных предметов.
Согласно исследованиям, женщины, выпивающие несколько стаканов пива или вина в неделю, имеют повышенный риск заболеть раком груди.
Синдром Шерешевского-Тёрнера | Симптомы и лечение синдрома Шерешевского-Тёрнера
^
- Новости
- Здоровье
- Семья и дети
- Питание и диеты
- Красота и мода
- Отношения
- Спорт
- О портале
Здоровье
- Диагностика
- Компьютерная томография
- МРТ диагностика
- Медицинские манипуляции
- Обследование организма
- Эндоскопия (эндоскопические исследования)
- Радионуклидная диагностика
- Рентген (рентгенологические исследования)
- Ультразвуковая диагностика (УЗИ)
- Лечение
- Операции
- Врачебные специальности
- Лечение болезней
- Обзор лекарственных средств
- Нетрадиционная медицина
- Стволовые клетки
- Физиотерапия
- Переливание крови
- Трансплантация
- Болезни
- Беременность, роды и послеродовой период
- Синдромы
- Хирургические болезни
- Болезни зубов (стоматология)
- Болезни молочных желез (маммология)
- Болезни суставов, мышц и соединительной ткани (ревматология)
- Рак (онкология)
- Болезни иммунной системы (иммунология)
- Болезни крови (гематология)
- Болезни сердца и сосудов (кардиология)
- Психическое здоровье (психиатрия)
- Травмы и отравления
- Болезни кожи и подкожной клетчатки (дерматология)
- Болезни легких, бронхов и плевры (пульмонология)
- Болезни уха, горла и носа (отоларингология)
- Болезни эндокринной системы и нарушения обмена веществ (эндокринология)
- Инфекции, передающиеся преимущественно половым путем (венерические болезни)
- Гинекологические болезни (гинекология)
- Болезни глаз (офтальмология)
- Болезни детей (педиатрия)
- Болезни печени и желчных путей
- Болезни прямой кишки и анальной области
- Болезни нервной системы (неврология)
- Болезни желудочно-кишечного тракта (гастроэнтерология)
- Инфекционные и паразитарные болезни
- Болезни мочеполовой системы
- Анализы
- Анализы на гормоны
- Гематологические исследования
- Генетические исследования
- Определение наркотиков
- Пренатальная диагностика
- ПЦР (Полимеразная цепная реакция, ПЦР-диагностика)
- Серологические исследования
- Токсикологические исследования
- Онкомаркеры
- Диагностика аут
Синдром Шерешевского — Тёрнера — это… Что такое Синдром Шерешевского — Тёрнера?
Синдром Шерешевского — Тёрнера
Синдром Шереше́вского — Тернера — хромосомная болезнь, сопровождающаяся характерными аномалиями физического развития, низкорослостью и половым инфантилизмом.
Основные сведения
Впервые эта болезнь как наследственная была описана в 1925 г. Н. А. Шерешевским, который считал, что она обусловлена недоразвитием половых желез и передней доли гипофиза и сочетается с врожденными пороками внутреннего развития. В 1938 г. Тернер выделил характерную для этого симптомокомплекса триаду симптомов: половой инфантилизм, кожные крыловидные складки на боковых поверхностях шеи и деформацию локтевых суставов. В России этот синдром принято называть синдромом Шерешевского — Тернера.
Четкой связи возникновения синдрома Тернера с возрастом и какими-либо заболеваниями родителей не выявлено. Однако беременности обычно осложняются токсикозом, угрозой выкидыша, а роды часто бывают преждевременными и патологическими. Особенности беременностей и родов, заканчивающихся рождением ребенка с синдромом Тернера, — следствие хромосомной патологии плода. Нарушение формирования половых желез при синдроме Тернера обусловлено отсутствием или структурными дефектами одной половой хромосомы (X-хромосомы).
У эмбриона первичные половые клетки закладываются почти в нормальном количестве, но во второй половине беременности происходит их быстрая инволюция (обратное развитие), и к моменту рождения ребенка количество фолликулов в яичнике по сравнению с нормой резко уменьшено или они полностью отсутствуют. Это приводит к выраженной недостаточности женских половых гормонов, половому недоразвитию, у большинства больных — к первичной аменорее (отсутствию менструаций) и бесплодию. Возникшие хромосомные нарушения являются причиной возникновения пороков развития. Возможно также, что сопутствующие аутосомные мутации играют определенную роль в появлении пороков развития, поскольку существуют состояния, сходные с синдромом Тернера, но без видимой хромосомной патологии и полового недоразвития.
При синдроме Тернера половые железы обычно представляют собой недифференцированные соединительнотканные тяжи, не содержащие элементов гонад. Реже встречаются рудименты яичников и элементы яичек, а также рудименты семявыносящего протока. Другие патологические данные соответствуют особенностям клинических проявлений. Наиболее важны изменения костно-суставной системы — укорочение пястных и плюсневых костей, аплазия (отсутствие) фаланг пальцев, деформация лучезапястного сустава, остеопороз позвонков. Рентгенологически при синдроме Тернера турецкое седло и кости свода черепа обычно не изменены. Отмечаются пороки сердца и крупных сосудов (коарктация аорты, незаращение боталлова протока, незаращение межжелудочковой перегородки, сужение устья аорты), пороки развития почек. Проявляются рецессивные гены дальтонизма и других заболеваний.
Синдром Шерешевского-Тёрнера встречается много реже, чем трисомия Х, синдром Клайнфельтера (ХХУ, ХХХУ), а так же ХУУ, что указывает на наличие сильного отбора против гамет, не содержащих половых хромосом, или против зигот ХО. Это предположение подтверждается достаточно часто наблюдемой моносомией Х среди спонтанно абортированных зародышей. В связи с этим допускается, что выжившие зиготы ХО являются результатом не мейотического, а митотического нерасхождения, или утраты X-хромосомы на ранних стадиях развития. Моносомии УО у человека не обнаружено. Популяционная частота 1:1500.
Клиническая картина и диагностика
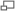 Складки кожи в области шеи — характерный признак болезни. На фото: девочка до и после пластической операции
Складки кожи в области шеи — характерный признак болезни. На фото: девочка до и после пластической операцииОтставание больных с синдромом Тернера в физическом развитии заметно уже с рождения. Примерно у 15 % больных задержка наблюдается в период полового созревания. Для доношенных новорожденных характерна малая длина (42—48 см) и масса тела (2500—2800 г и менее). Характерными признаками синдрома Тернера при рождении являются избыток кожи на шее и другие пороки развития, особенно костно-суставной и сердечно-сосудистой систем, «лицо сфинкса», лимфостаз (застой лимфы, клинически проявляющийся крупными отеками). Для новорожденного характерны общее беспокойство, нарушение сосательного рефлекса, срыгивание фонтаном, рвота. В раннем возрасте у части больных отмечают задержку психического и речевого развития, что свидетельствует о патологии развития нервной системы. Наиболее характерным признаком является низкорослость. Рост больных не превышает 135—145 см, масса тела часто избыточна. У больных с синдромом Тернера патологические признаки по частоте встречаемости распределяются следующим образом: низкорослость (98 %), общая диспластичность (неправильное телосложение) (92 %), бочкообразная грудная клетка (75 %), укорочение шеи (63 %), низкий рост волос на шее (57 %), высокое «готическое» нёбо (56 %), крыловидные складки кожи в области шеи (46 %), деформация ушных раковин (46 %), укорочение метакарпальных и метатарзальных костей и аплазия фаланг (46 %), деформация локтевых суставов (36 %), множественные пигментные родинки (35 %), лимфостаз (24 %), пороки сердца и крупных сосудов (22 %), повышенное артериальное давление (17 %).
Половое недоразвитие при синдроме Тернера отличается определенным своеобразием. Нередкими признаками являются геродермия (патологическая атрофия кожи, напоминающая старческую) и мошонкообразный вид больших половых губ, высокая промежность, недоразвитие малых половых губ, девственной плевы и клитора, воронкообразный вход во влагалище. Молочные железы у большинства больных не развиты, соски низко расположены. Вторичное оволосение появляется спонтанно и бывает скудным. Матка недоразвита. Половые железы не развиты и представлены обычно соединительной тканью. При синдроме Тернера отмечается склонность к повышению артериального давления у лиц молодого возраста и к ожирению с нарушением питания тканей.
Интеллект у большинства больных с синдромом Тернера практически сохранен, однако частота олигофрении все же выше. В психическом статусе больных с синдромом Тернера главную роль играет своеобразный психический инфантилизм с эйфорией при хорошей практической приспособляемости и социальной адаптации.
Диагноз синдрома Тернера основывается на характерных клинических особенностях, определении полового хроматина (вещества клеточного ядра) и исследовании кариотипа (хромосомного набора). Дифференциальный диагноз проводится с нанизмом (карликовостью), для исключения которого проводится определение содержания гормонов гипофиза в крови, особенно гонадотропинов.
Лечение
На первом этапе терапия заключается в стимуляции роста тела анаболическими стероидами и другими анаболическими препаратами. Лечение следует проводить минимальными эффективными дозами анаболических стероидов с перерывами при регулярном гинекологическом контроле. Главным видом терапии больных является эстрогенизация (назначение женских половых гормонов), которую следует проводить с 14-16 лет. Лечение приводит к феминизации телосложения, развитию женских вторичных половых признаков, улучшает трофику (питание) половых путей, уменьшает повышенную активность гипоталамо-гипофизарной системы. Лечение следует проводить в течение всего детородного возраста больных. При синдроме Тернера у мужчин применяется заместительная терапия мужскими половыми гормонами.
Прогноз для жизни при синдроме Тернера благоприятный, исключение составляют больные с тяжелыми врожденными пороками сердца и крупных сосудов и почечной гипертензией. Лечение женскими половыми гормонами делает больных способными к семейной жизни, однако абсолютное большинство из них остаются бесплодными.
Кроме этого в последнее время проводится терапия соматотропином или человеческим гормоном роста
Проблемы долгосрочного лечения больных синдромом Шерешевского–Тернера | #03/10
Синдром Шерешевского–Тернера (СШТ) — генетически детерминированное заболевание, развивающееся в результате качественного либо количественного нарушения одной половой Х-хромосомы. По данным разных авторов частота встречаемости заболевания колеблется от 1:2000 до 1:2500 новорожденных девочек. Классическое описание СШТ было сделано американским врачом Генри Тернером в 1938 году. Однако еще раньше, 12 ноября 1925 года, профессор Н. А. Шерешевский представил клинические данные женщины 25 лет: низкий рост (132 см), выраженный половой инфантилизм — первичная аменорея, отсутствие вторичных половых признаков, недоразвитие внутренних гениталий.
Основными задачами лечения больных с СШТ в детском и подростковом возрасте являются увеличение конечного роста, формирование вторичных половых признаков с установлением регулярного менструального цикла, коррекция пороков развития и профилактика остеопороза. Своевременно начатая и адекватно проводимая терапия обеспечивает в последующем женщинам с СШТ полноценную активную жизнь. Однако многообразие клинических проявлений заболевания, неоднородность патофизиологических механизмов их развития создают проблемы в достижении оптимальных результатов лечения.
Наряду с низкорослостью и гипергонадотропным гипогонадизмом у больных с СШТ часто наблюдаются пороки развития внутренних органов, обусловленные хромосомными аберрациями. По мнению некоторых авторов, хромосомные аномалии, характерные для СШТ, могут иметь до 3% всех эмбрионов женского пола, однако вследствие спонтанных абортов лишь 1% плодов сохраняется до конца беременности. Таким образом, СШТ ответственен за 7–10% всех самопроизвольных прерываний беременности [1].
Гипогонадизм встречается почти у 98% больных СШТ. Редко диагностируют сохранную функцию яичников, проявляющуюся, как правило, увеличением молочных желез. Для первичной овариальной недостаточности, развивающейся в результате дисгенезии гонад, характерно значительное увеличение уровня гонадотропных гормонов в результате отсутствия отрицательного подавляющего влияния половых гормонов на гипоталамо-гипофизарную систему. У девочек с СШТ уже в первые недели жизни определяется повышение уровня гонадотропных гормонов, преимущественно фолликулостимулирующего гормона (ФСГ). Такое повышение сохраняется до двухлетнего возраста. В возрасте от 2 до 6 лет происходит снижение уровня гонадотропных гормонов в результате активации универсального центрального механизма, подавляющего секрецию гонадотропин-релизинг-гормона (ЛГ-РГ) независимо от уровня половых гормонов. В возрасте 5–6 лет начинают вновь повышаться ФСГ и лютеинизирующий гормон (ЛГ). В период полового развития отмечено увеличение уровня гонадотропных гормонов (ФСГ, ЛГ) в 10 раз [1, 3, 4, 14].
У 5–7% девочек с гипергонадотропным гипогонадизмом может отмечаться спонтанное половое развитие, чаще при мозаичном варианте хромосомного набора. У этих девочек спонтанное половое развитие чаще является неполным и не приводит к нормальной и длительной овариальной функции. Однако в литературе описаны случаи спонтанных и повторных беременностей и родов у женщин с СШТ. Необходимо отметить, что проявлением истинного пубертата следует считать только увеличение молочных желез. Вторичное оволосение — лобковое и аксиллярное — развивается спонтанно у всех девочек с гипергонадотропным гипогонадизмом к 12–13 годам под влиянием надпочечниковых андрогенов [1, 3].
Больные с СШТ в 100% случаев имеют низкий рост. Девочки уже при рождении имеют низкую массу и длину тела. В течение первых 2–3 лет скорость роста достаточно стабильна. В последующем в возрасте от 3 до 11 лет отмечается выраженное снижение темпов роста. В возрасте пубертата ростового скачка не происходит, и отставание в росте становится максимальным [1, 14, 22, 23]. До недавнего времени патогенез низкорослости при СШТ оставался неясным.
Существовал ряд гипотез, объясняющих этот симптом заболевания. При этом влияние различных вариантов кариотипа при СШТ на конечный рост доказано не было. В последнее десятилетие было установлено, что ведущую роль в патогенезе низкорослости у пациентов с гипергонадотропным гипогонадизмом имеют непосредственные генетические нарушения, обусловленные делецией гена SHOX. Это ген, локализованный в псевдоаутосомной области на конце короткого плеча Х-хромосомы, с которым связывают низкорослость при СШТ и некоторых других синдромах, сопровождающихся мезомиелической дисплазией костной ткани. Ген SHOX (short stature homeobox gene) кодирует белок, содержащий гомеодомен, который с наибольшей вероятностью работает как регулятор транскрипции. Снижение экспрессии SHOX-гена может быть ассоциировано не только с низкорослостью, но и с деформацией скелета (деформация Маделунга, вальгусная деформация, микрогнатия, «готическое небо», укорочение конечностей и пястных костей) [3].
Пациенты с СШТ требуют не только коррекции гонадной недостаточности, но и стимуляции роста. Выраженная задержка роста начинает формироваться у детей с 5–6 лет, и конечный рост колеблется около 140 см.
Соматотропная функция у больных с СШТ на фоне стимуляционных проб продемонстрировала адекватный выброс соматотропного гормона (СТГ) в ответ на стимуляцию. У девочек с СШТ снижение секреции СТГ до периода полового развития происходит в результате дефицита эстрогенов. У пациентов также было обнаружено снижение уровня инсулиноподобного фактора роста-1 (ИФР-1), необходимого для осуществления периферического действия соматотропного гормона [1, 2, 5, 6]. Некоторые ученые полагают, что у больных с гипергонадотропным гипогонадизмом отмечается частичная резистентность к гормону роста и ИФР-1. Однако это требует дальнейшего изучения.
В мировой практике в настоящее время начато широкое применение рекомбинантных препаратов гормона роста у девочек с СШТ. Многочисленные исследования показали, что препараты соматропина ускоряют рост у этих детей и увеличивают их конечное значение до 150–155 см. Увеличение конечного роста в результате лечения супрафизиологическими дозами соматропина по данным разных авторов весьма вариабельно — от 3 до 16 см по отношению к прогнозируемому без лечения росту [14, 22, 25, 26]. В связи с тем, что спонтанный пубертат развивается лишь у 20% девочек с СШТ, возникает необходимость индукции полового развития с помощью эстрогенов. При этом эстрогены активируют закрытие эпи
Синдром Тёрнера (Синдром Шерешевского — Тёрнера) [LifeBio.wiki]
Синдром Тернера, также известный как синдром Ульриха-Тернера, дизгенез гонад и 45, X, представляет собой состояние, при котором женщина частично или полностью утрачивает X-хромосому. 1) Признаки и симптомы варьируются среди страдающих заболеванием. Низкая линия роста волос позади шеи, низкая линия роста волос позади шеи.Обычно у страдающих синдромом отсутствуют периоды менструации, не развиваются молочные железы, они не способны иметь детей. Пороки сердца, сахарный диабет и низкий уровень тиреоидного гормона встречаются чаще. Большинство людей с синдромом Тернера имеет нормальные умственные способности. Многие, тем не менее, имеют проблемы с пространственным представлением, с математикой. Часто возникают проблемы со слухом и зрением. 2) Синдромнера обычно не наследуется от родителей. 3) Отсутствуют известные факторы риска окружающей среды, возраст матери не играет роли.Синдромнера связана с хромосомной аберрацией, при которой вся или часть одной из X-хромосом утрачивается или меняется. В то время как большинство людей имеет 46 хромосом, люди с синдромом Тернера, как правило, имеют всего 45. Хромосомная аберрация может быть представлена только в некоторых клетках, в данном случае состояние носит название синдром Тернера с мозаицизмом. В случаях симптомов обычно меньше или, возможно, они вовсе отсутствуют. Диагностика основывается на физических признаках и генетическом тестировании. 4) Лечения синдрома Тернера не существует. Тем не менее, лечение может облегчить симптомы. Инъекции человеческого гормона роста в детстве могут увеличить взрослый рост. Терапия за ущерб эстрогена может вызвать развитие молочных желез и таза. Часто требуется медицинский уход с целью управления проблемами со здоровьем, с помощью связывается синдром Тернера. Распространенность синдрома Тернера составляет от 1 на 2000 до 1 на 5000 женщин при рождении. 5) Все регионы мира и культуры подвержены заболеванию практически в равной степени.Люди с синдромом имеют более короткую среднюю продолжительность жизни, проблемы с сердцем и сахарным диабетом. Генри Тернер первый описал состояние в 1938 г. В 1964 г. было выявлено, что синдром связан с хромосомной аберрацией.
Признаки и симптомы
Далее перечень обычных симптомов синдрома Тернера. Важно заметить, что субъект может иметь любую комбинацию симптомов и маловероятно имеет все симптомы.
Низкий рост
Лимфедема (опухание) рук и стоп
Широкая грудь (щитовая грудь) и широко расставленные соски
Низкая линия роста волос
Низко посаженные уши
Бесплодие
Рудиментарные яичники в виде гонадного слоя (недоразвитые гонадальные структуры, которые позже становятся фиброзными)
Аменорея, или отсутствие периодов менструации
Повышенный вес, ожирение
Щитообразный торакс сердца
Укороченная пясть IV
Маленькие ногти на пальцах руки
Характерные лицевые особенности
Перепончатая шея в результате шейной гигромы в младенчестве
Стеноз аортального клапана
Коарктация аорты
Двустворчатый аортальный клапан
Подковообразная почка
Зрительные нарушения склеры, роговицы, глаукома и т.д.
Ушные инфекции и потеря слуха
Высокое соотношение талия-бедра (бедра не намного больше, чем талия)
Синдром дефицита внимания с гиперактивностью, или СДВГ (проблемы с концентрацией, памятью, вниманием, при этом гиперактивность наблюдается в основном у детей и подростков)
Нарушение невербальной обучаемости (проблемы с математикой, социальными навыками и пространственными соотношениями)
Другие особенности направленная маленькая нижняя челюсть (микрогнатия), вальгусную деформацию локтевого сустава, 6) мягкие изогнутые ногти, ладонную складку и птоз верхнего века.Реже встречаются пигментные невусы, потеря слуха и подъем неба (узкая верхняя челюсть). Синдромнера сам по себе проявляется по-разному у каждой женщины, подверженной заболеванию. В то время как большая часть нарушений безвредна, с синдромом связаны значительные медицинские проблемы.
Пренатальные
Несмотря на очень хороший постнатальный прогноз, 99% случаев зачатий с синдромом окончание выкидышем или мертворождением, при этом до 15% всех выкидышей кариотип 45, X.Среди случаев, которые обнаруживаются регулярным амниоцентезом или биопсией ворсин хориона, одно исследование выявило, что распространенность среди синдрома Тернера среди протестированных случаев беременности было в 5,58 и 13,3 раза выше, чем здоровых младенцев той же группы людей. 7)
Сердечно-сосудистые
Распространенность аномалий сердечно-сосудистой системы
Распространенность аномалий сердечно-сосудистой системы пациентов с синдромом Тернера находится в диапазоне от 17% (Ландин-Вильгельмсен и др., 2001) до 45% (Доусон-Фолк и др., 1992). Изменчивость, обнаруженная в различных исследованиях, главным образом объясняется вариациями неинвазивных методов, применяют для анализа и типы повреждений, которые они описывают (Го и др., 2004). Тем не менее, Сиберт, 1998 г., свидетельствует, что это может объясняться всего лишь небольшим числом субъектов в большинстве исследований. Различные кариотипы могут давать различную распространенность аномалий сердечно-сосудистой системы. Два исследования распространенной аномалий сердечно-сосудистой системы в 30% и 38% 8) в группе 45, X моносом чистой.Но, учитывая кариотипы других групп, они сообщили о распространенности в 24,3% и 11% у пациентов с мозаичной X моносомией и распространенности в 11% у пациентов с X хромосомными аномалиями строения. Более высокая распространенность в группе чистой 45, X моносомии в первую очередь объединяет со специалистами Clips разницей в распространенности аномалий аортального и коарктации клапана, двух наиболее частых аномалий сердечно-сосудистой системы.
Врожденный порок сердца
Наиболее часто наблюдаются врожденные обструктивные повреждения левой части сердца, что ведет к кровотоку в этой части сердца.Они охватывают двустворчатый аортальный клапан и коарктацию (сужение) аорты. Сиберт, 1998 г., обнаружила, что более 50% аномалий сердечно-сосудистой системы, наблюдаемых в ее исследовании субъектов с синдромом Тернера, составляющими двустворчатые аортальные клапаны и коарктация аорты, в отдельности или в сочетании. Другие врожденные аномалии сердечно-сосудистой системы, такие как частичный аномальный венозный отток и стеноз аортального клапана или регургитация аорты, чаще встречаются у страдающих синдромом Тернера, чем у общей совокупности населения.Гипоплазия левой половины сердца представляет собой наиболее серьезное сокращение левосторонних структур.
Двустворчатый аортальный клапан
До 15% взрослых с синдромом Тернера двустворчатые аортальные клапаны, означающие, имеют только две, вместо трех, части клапанов основного кровеносного сосуда, идущего от сердца. Используется двустворчатые клапаны надлежащим образом регулировать кровоток, данное состояние может быть не обнаружено в отсутствии регулярного обследования.Тем не менее, двустворчатые клапаны более вероятно изнашиваются и продолжают функционировать. Также возникает кальцификация клапанов, которая может привести к прогрессирующей клапанной дисфункции, что вызывает стеноз аорты и регургитацию. 9) Распространенность от 12,5% до 17,5% (Доусон-Фолк и др., 1992), двустворчатый аортальный клапан представляет собой наиболее часто наблюдаемую врожденную аномалию, влияющую на сердце при данной синдроме. Обычно возникает в отдельной, но может наблюдаться в сочетании с другими аномалиями, в частности, коарктацией аорты.
Коарктация аорты
От 5% до 10% рожденных с синдромом Тернера имеют коарктацию аорты, врожденное сужение нисходящей части аорты, обычно удаленной от левой подключичной артерии (артерия, которая ответвляется от дуги аорты к левой руке) и расположенной протока (также называется «юкстадактальная»). Расчетные показатели распространенности данной аномалии среди пациентов с синдромом Тернера находятся в диапазоне от 6,9% до 12,5%. Коарктация аорты у женщин свидетельства о синдроме Терминал и о потребности в дальнейших анализах, таких как кариотип.
Частичный аномальный венозный отток
Данная аномалия представляет собой сравнительное врожденное заболевание сердца по отношению к общей совокупности населения. Распространенность примерно данной аномалии также является низкой (2,9%) при синдроме Тернера. Тем не менее, сравнительный риск составляет 320 по сравнению с общей совокупностью населения. Что странно, синдром Тернера связывается с необычными формами частичного аномального венозного оттока. 10) Пациенты с повышенной предрасположенностью к бактериально-эндокардиту приводят к повышенной предрасположенности к бактериально-эндокардиту.Следовательно, когда выполняются процедуры с высоким риском развития эндокардита, такие как профессиональная чистка зубов, для профилактики должны приниматься антибиотики. Синдромнера часто связывается с устойчивой гипертензией, в некоторых случаях в детстве. У пациентов с синдромом пациента нет пациентов с синдромом Тернера и гипертензией специфическая причина. В остальном она обычно связана с сердечно-сосудистыми заболеваниями или почечными аномалиями, включая коарктацию аорты.
Дилатация, расслоение и разрыв аорты
Два исследования свидетельствуют о дилатации аорты при синдроме Тернера, обычно охватывающей основание восходящей части дуги аорты и изредка распространяющейся через дугу на нисходящую часть аорты, либо на место предыдущей коарктации зажившей аорты. 11)
Аллен и др., 1986 г., которые изучали 28 девочек с синдромом Тернера, показали значительно больший средний диаметр основания аорты у пациентов с синдромом Тернера, чем у контрольной группы (оценивался по отношению к площади поверхности тела). Тем не менее, диаметр основания аорты, обнаруженный у пациентов с синдромом Тернера, все жеился в диапазоне предельных значений.
Это было подтверждено исследованием Доусона-Фолка и др., 1992 г. которые, изучали 40 пациентов с синдромом Тернера. Они представили в основном аналогичные результаты: больший средний диаметр основания аорты, который же все остается в пределах нормального диапазона в отношении площади поверхности тела.
Сиберт, 1998 г., обращает внимание на недоказанность, что диаметр основания аорты, который сравнительно велик в отношении площади поверхности тела, но все же остается в пределах нормального диапазона, предполагает риск прогрессирующей дилатации. 12)
Распространенность аортальных аномалий
Распространенность дилатации основания аорты у пациентов с синдромом Тернера находится в диапазоне от 8,8% до 42%. Даже если каждая дилатация основания аорты обязательно прогрессируют до расслоения аорты (кольцевой или поперечный разрыв интимы), могут возникать осложнения, такие как расслоение и разрыв аорты, которые приводят к смерти. Известно, что это связано с расслоением и разрывом аорты.Расслоению аорты подвержены от 1% до 2% пациентов с синдромом Тернера. Как результат, дилатация основания аорты должна быть серьезно принята во внимание, поскольку она может привести к фатальному расслоению аорты. Настоятельно рекомендуется регулярное обследование.
Факторы риска в отношении разрыва аорты
Хорошо известно, что аномалии сердечно-сосудистой системы (обычно двустворчатый аортальный клапан, коарктация аорты и некоторые другие побочные сердечно-сосудистые аномалии) и гипертензия предрасполагают к дилатации и расслоению аорты у общей совокупности населения.В то же время выявлено, что это факторы при синдроме Тернера. Более того, аналогичные факторы риска были обнаружены более чем 90% пациентов с синдромом Тернера, у которых улучшилась дилатация аорты. Только небольшое число пациентов (примерно 10%) не имеют явно предрасполагающих факторов риска. Важно отметить, что риск гипертензии в 3 крат выше у пациентов с синдромом Тернера. По причине связи с расслоением аорты, кровяное давление должно постоянно контролироваться, а гипертензия должна подвергаться агрессивному лечению с целью поддержания кровяного давления ниже 140/80 мм рт.ст. Было выявлено, что, как и в случае других аномалий сердечно-сосудистой системы, осложнения дилатации аорты в достаточной степени связаны с кариотипом 45, X.
Патогенез расслоения и разрыва аорты
Точная роль, данные факторы риска играют в процессе, ведущем к таким фатальным осложнениям, до сих пор достаточно не ясна. Дилатация основания предположительно связана с мезенхимальным дефектом как патологическим свидетельством кистозного медионекроза аорты, обнаруженными территориями.Связь между аналогичным дефектом и дилатацией аорты точно при таком состоянии как синдром Марфана. Кроме того, аномалии в других мезенхимальных тканях (костный матрикс и лимфатические сосуды) свидетельствуют об аналогичном исходном мезенхимальном дефекте у пациентов с синдромом Тернера. У пациентов с синдромом Тернера степени более высокий риск дилатации и расслоения аорты в отсутствии предрасполагающих факторов.Таким образом, риск расслоения аорты при синдроме является результатом аномалий сердечно-сосудистой системы и гемодинамических факторов риска, чем отражением наследственной аномалии соединительной ткани (Сиберт, 1998). Течение развития дилатации основания аорты не известно, но поскольку она имеет летальный потенциал, аортальная аномалия должна тщательно обследоваться.
Скелетные
Нормальное скелетное развитие ингибируется в связи с широким множеством факторов, главным образом, гормональными.Средний рост женщины с синдромом Тернера, отсутствие лечения гормоном роста, составляет 4 фута 7 дюймов (140 см). Пациенты с мозаицизмом Тернера могут достичь нормальный средний рост. Четвертая метакарпальная кость (четвертый палец ноги и безымянный палец) может быть необычно короткой, как и пятая. В связи с нарушенной выработкой эстрогена, у многих страдающих синдромом Тернера развивается остеопороз. Это в дальнейшем может уменьшать рост, а также плохить искривление позвоночника, что другое к сколиозу.Он также связывается с повышенным риском переломов костей.
Почки
Приблизительно одна треть всех женщин с синдромом Тернера имеет одну из трех аномалий почек:
Единственная подковообразная почка с одной стороны организма.
Атипичная система сбора мочи.
Плохой кровоток, идущий к почкам.
Некоторые из состояний могут быть исправлены хирургически.Даже при наличии данных аномалий почки женщин с синдромом Тернера могут нормально функционировать. Тем не менее, как отмечалось выше, проблемы с почками могут быть связаны с гипертензией.
Щитовидная железа
Приблизительно одна треть всех женщин с синдромом Тернера имеет заболевание щитовидной железы. Обычно оно представлено гипотиреозом, а именно тиреоидитом Хашимото. Когда обнаружен, он может легко поддаваться лечению добавками тиреоидного гормона.
Сахарный диабет
Женщины с синдромом Тернера имеют незначительно повышенный риск развития сахарного диабета типа 1 в детстве и в степени повышенного риска развития сахарного диабета типа 2 во взрослом возрасте.Риск развития сахарного диабета типа 2 может быть снижен за счет поддержания здорового веса.
Когнитивная функция
Синдром Тернера обычно не вызывает умственной отсталости или нарушения когнитивной функции. Тем не менее, сложности обучения распространены среди женщин с синдромом Тернера, в частности, специфическая сложность понимания пространственных связей, а также нарушение способности к невербальному обучению. Также может он проявляться как сложность с двигательным контролем или математикой.В то время как не поддается лечению, в большинстве случаев это не вызывает трудностей в повседневной жизни. Основные пациенты с синдромом Тернера нанимаются на работу как полноценные люди и ведут продуктивную жизнь. Также существует редкая разновидность синдрома Тернера, известная как «Кольцевой-X синдром Тернера», который имеет примерно 60-процентную связь с умственной отсталостью. Данная разновидность охватывает 2–4% всех случаев синдрома Тернера. 13)
Репродуктивная система
Женщины с синдромом Тернера практически в всех случаях бесплодны.В то время как некоторые женщины с синдромом могут успешно зачать ребенка и перенести беременность, это достаточно редко и, как правило, ограничивается теми женщинами, чей кариотип не соответствует 45, X. Даже когда такая беременность возникает, превышает средний риск выкидыша или врожденных дефектов, включая синдромы Тернера Дауна. Некоторые женщины с синдромом Тернера, неспособные зачать без медицинского вмешательства, использовать экстракорпоральное оплодотворение или другие средства преодоления бесплодия. 14) Терапия за ущерб эстрогена, как правило, используется как средство стимулирования ростковых половых признаков тогда, когда должно начаться половое созревание. В то время как достаточно небольшое число женщин с синдромом Тернера менструируют самопроизвольно, эстрогеновая терапия требует регулярного выведения оболочки оболочки матки с целью предотвращения ее чрезмерного роста. Кровотечение ежемесячно, как менструация, ежемесячно, ежемесячно, по желанию пациента.Эстрогеновая терапия не делает женщину с нефункциональными яичниками способной к деторождению, но играет роль в искусственном оплодотворении; здоровье матки необходимо поддерживать с помощью эстрогена, если пригодная для оплодотворения женщина с синдромом Тернера желает использовать экстракорпоральное оплодотворение (с использованием донорских ооцитов). Синдром Тернера представляет собой причину первичной аменореи, синдрома истощения яичников (гипергонадотропный гипогонадизм), полосковидных гонад и бесплодия.Невозможность развития вторичных половых признаков (сексуальный инфантилизм) является типичной для состояния. Рекомендуется гонадэктомия, в особенности в проблемных случаях синдрома яичникового синдрома. Синдром Тернера характеризуется первичной аменореей, синдромом истощения яичников, полосковидными гонадами и родием. Тем не менее, технологии (особенно донорство ооцитов) обеспечивают данным пациентам возможность зачать ребенка.Было принято решение, что беременность может нести риск сердечно-сосудистых заболеваний матери. Более того, несколько исследований о повышенном риске расслоения аорты при беременности. Сообщалось даже о трех случаях летального исхода. Влияние эстрогена подвергалось исследованиям, но остается неясным. Предполагается, что высокий риск расслоения аорты во время беременности с синдромом Тернера может быть скорее с повышенной гемодинамической нагрузкой, чем с высоким уровнем эстрогена.Безусловно, данные результаты важны и должны приниматься во внимание в отношении беременных пациентов с синдромом Тернера.
Причина
Синдром Тернера представляет собой последствие отсутствия двух полных копий X-хромосомы в некоторых во всех во всех клетках. Атипичные клетки могут иметь только одну хромосому X (45, X) или могут быть подвержены одному из нескольких типов частной моносомии, как ситуация короткой p дуги одной X-хромосомы (46, X, дел (Xp) или изохромосомы с двумя q дугами (46, X, i (Xq).У мозаичных субъектов клетки с X-моносомией (45, X) могут наблюдаться наряду с нормальными клетками (46, XX), клетками, которые имеют частичную моносомию, или клетками, которые имеют Y-хромосому (46, XY). Наличие мозаицизма, по расчетам пациентов, сравнительно часто встречается у подверженных синдрому Тернера (67–90%). 15)
Наследование
В большинстве случаев, когда возникает моносомия, X-хромосома берется от матери. Это может быть связано с нерасхождением у отца.Также у отцов часто появляются мейотические ошибки, которые ведут к образованию хромосомы X с помощью дуги и патологической Y-хромосомы. Изохромосома X или кольцевая хромосома X, с другой стороны, формируются с равной встречаемостью у обоих родителей. В целом, функциональная X-хромосома в большинстве случаев исходит от матери. В большинстве случаев синдром синдром представляет собой случайное событие, и для родителей ребенка с синдромом Тернера риск повторения для беременностей не повышается.Редкие исключения могут быть сбалансированной транслокации X-хромосомы у родителя, либо случаев, в которых мать имеет 45, X мозаицизм, ограниченный ее первичными половыми клетками. 16)
Диагностика
Пренатальная
Синдром Тернера может быть диагностирован во время беременности с помощью амниоцентеза или биопсии ворсин хориона. Как правило, плод с синдромом Тернера может быть выявлен посредством отклонения от нормы результатов ультразвукового исследования (например, порок сердца, аномалия почек, шейная гигрома, асцит).В исследовании 19 европейских журналов учета, 67,2% пренатально диагностированных случаев синдрома Тернера были обнаружены за счет аномалий при ультразвуковом исследовании. В 69,1% была представлена одна аномалия, а в 30,9% случаев — две и более аномалии. Повышенный риск развития синдрома Тернера также может быть выявлен с помощью отклонения от нормы триады или квадруплета в обследовании материнской крови. Плод, диагностированный посредством положительного анализа материнской сыворотки, чаще имеет мозаичный кариотип, чем тот, диагноз которого основан на аномалиях ультразвукового исследования, и, наоборот, субъекты с мозаичными кариотипами, вероятно, имеют связанные ультразвуковые аномалии.Хотя риск рецидива не повышается, семьям, в которых имеется случай беременности или с синдромом Тернера, рекомендуется генетическое консультирование.
Постнатальная
Синдром Тернера может быть диагностирован постнатально в любом возрасте. Часто он диагностируется при рождении в связи с проблемами сердца, необычно широкой шеей и отеком рук и стоп. Тем не менее, часто встречается, что он остается недиагностированным в течение нескольких лет, пока девочка не достигла возраста полового созревания / пубертатного возраста и обычно не может начать развиваться надлежащим образом (изменения, связанные с половым созреванием, не произут).В детстве о синдроме Тернера может свидетельствовать низкий рост. Анализ, носящий название кариотип или анализ хромосом, анализирует хромосомный состав. Это предпочтительный анализ с целью диагностики синдрома Тернера.
Лечение
Синхронизация Тернера представляет собой хромосомное состояние, лечение не существует. Тем не менее, для облегчения симптомов может быть предпринято множество мер. Например: 17)
Врачи могут использовать инъекцию гормона роста, известного как Генотропин («Pfizer»)
- Гормон роста, в отдельной или низкой дозе андрогена, увеличит рост и, возможно, приведет к достижению взрослого роста.Гормон роста утвержден. Управление по контролю за продуктами питания и лекарственными средствами США для лечения синдрома. Существуют доказательства, что он эффективен даже у только начавших детей. 18)
Терапия за ущерб эстрогена, как пероральные противозачаточные средства, применялась с тех пор, когда такое состояние было описано в1938 г., с целью развития вторичных половых признаков. Эстрогены крайне важны для обеспечения целостности костей, здоровья сердечно-сосудистой системы и тканей.Женщины с синдромом Тернера, не демонстрирующие спонтанного полового созревания и не проходящие лечение с помощью эстрогена, находятся в группе повышенного риска развития остеопороза и сердечных заболеваний.
Современные репродуктивные технологии также применяются с целью помочь женщинам с синдромом Тернера в желании зачать ребенка. Например, для образования эмбриона может вынашиваться женщиной с синдромом Тернера.
Маточная зрелость положительно связана с годами применения эстрогена, историей спонтанного менархе и отрицательно связана с отсутствием текущей терапии за ущерб гормона.
Эпидемиология
Примерно в 99 процентов зачатия при синдроме Тернера происходит самопроизвольное прерывание во время первого триместра. 19) Синдром Тернера охватывает 10 процентов от общего числа выкидышей в США.Встречаемость синдрома Тернера у рождающихся здоровых девочек предположительно составляет 1 на 2000 год.
История
Синдром получил название в честь Генри Тернера, эндокринолога из Иллинойса, который описал состояние в 1938 г. 20) В Европе часто называют синдромом Ульриха-Тернера или даже синдром Бонневи-Ульриха-Тернера, признавая, что ранние случаи синдрома также были развитыми европейскими врачами. Первое опубликованное сообщение о женщине с кариотипом 45, X было сделано в 1959 г.доктором Чарльзом Фордом и его коллегами в Харвелле, Оксфордшир и госпитале Гая в Лондоне. [44] Он был обнаружен у 14-летней девочки с признаками синдрома Тернера.
: Теги
Читать еще: Бендамустин (Рибомустин), Каркаде (Суданская роза), Наттокиназа, Никотин, Остролодочник серповидный,
Список использованной литературы:
1) «Синдром Тернера: Обзор». Юнис Кеннеди Шрайвер Национальный институт детского здоровья и развития человека.3 апреля 2013 г. Проверено 15 марта 2015 г. 2) Sybert VP, McCauley E; Макколи (сентябрь 2004 г.). «Синдром Тернера». N. Engl. J. Med. 351 (12): 1227–38. DOI: 10.1056 / NEJMra030360. PMID 15371580. 3) «Сколько людей пострадали или находятся в группе риска?». Юнис Кеннеди Шрайвер Национальный институт детского здоровья и развития человека. 30 ноября 2012 г. Проверено 15 марта 2015 г. 4) «Как поставщики медицинских услуг диагностируют синдром Тернера?».Юнис Кеннеди Шрайвер Национальный институт детского здоровья и развития человека. 30 ноября 2012 г. Проверено 15 марта 2015 г. 5) Марино, Брэдли С. (2013). Чертежи педиатрии (изд. Шестое. Ред.). Филадельфия: Уолтерс Клувер / Липпинкотт Уильямс и Уилкинс. п. 319. ISBN 9781451116045. 6) Глава об аменорее в: Bradshaw, Karen D .; Schorge, John O .; Шаффер, Джозеф; Лиза М. Халворсон; Хоффман, Барбара Г. (2008). Гинекология Уильямса.McGraw-Hill Professional. ISBN 0-07-147257-6. 7) Гравхольт С.Х., Юул С., Наераа Р.В., Хансен Дж.; Юул; Наэра; Хансен (1996-01-06). «Пренатальная и постнатальная распространенность синдрома Тернера: регистрационное исследование». BMJ (Издание клинических исследований) 312 (7022): 16–21. DOI: 10.1136 / bmj.312.7022.16. PMC 2349728. PMID 8555850. 8) Gøtzsche CO, Krag-Olsen B., Nielsen J, Sørensen KE, Kristensen BO; Краг-Ольсен; Нильсен; Соренсен; Кристенсен (ноябрь 1994 г.). «Распространенность сердечно-сосудистых пороков развития и ассоциация с кариотипами при синдроме Тернера».Arch Dis Child. 71 (5): 433–6. DOI: 10.1136 / adc.71.5.433. PMC 1030059. PMID 7826114. 9) Эльшейх М., Дангер Д. Б., Конвей Г. С., Васс Дж. А.; Дангер; Конвей; Васс (февраль 2002 г.). «Синдром Тернера в зрелом возрасте». Endocr. Откровение 23 (1): 120–40. DOI: 10.1210 / er.23.1.120. PMID 11844747. 10) Прандстраллер Д., Маццанти Л., Пиччио Ф.М., Маньяни С., Бергамаски Р., Перри А., Цингос Е., Каччиари Е. Маццанти; Пиччио; Маньяни; Бергамаски; Перри; Цинго; Cacciari (1999).«Синдром Тернера: кардиологический профиль в соответствии с различными хромосомными паттернами и долгосрочное клиническое наблюдение 136 пациентов, не прошедших предварительный отбор». Педиатр Кардиол 20 (2): 108–12. DOI: 10.1007 / s0024696. PMID 9986886. 11) Лин А.Е., Липпе Б., Розенфельд Р.Г.; Липпе; Розенфельд (июль 1998 г.). «Дальнейшее определение дилатации, расслоения и разрыва аорты у пациентов с синдромом Тернера». Педиатрия 102 (1): e12. DOI: 10.1542 / peds.102.1.e12. PMID 9651464. 12) Sybert VP (январь 1998 г.).«Сердечно-сосудистые мальформации и осложнения при синдроме Тернера». Педиатрия 101 (1): E11. DOI: 10.1542 / peds.101.1.e11. PMID 9417175. 13) Берковиц Дж., Стамберг Дж., Плотник Л. П., Лейнс Р; Стамберг; Плотник; Lanes (июнь 1983 г.). «Пациенты с синдромом Тернера с кольцевой Х-хромосомой». Clin Genet. 23 (6): 447–53. DOI: 10.1111 / j.1399-0004.1983.tb01980.x. PMID 6883789. 14) Ховатта О. (1999). «Беременности у женщин с синдромом Тернера». Анналы медицины 31 (2): 106–10.DOI: 10.3109 / 07853899908998785. PMID 10344582. 15) Креспи Б (2008). «Синдром Тернера и эволюция полового диморфизма человека». Эволюционные приложения 1 (3): 449–461. DOI: 10.1111 / j.1752-4571.2008.00017.x. 16) Фриас Дж. Л., Давенпорт М. Л.; Давенпорт; Комитет по генетике, секция эндокринологии (2003). «Наблюдение за здоровьем детей с синдромом Тернера». Педиатрия 111 (3): 692–702. DOI: 10.1542 / педс.111.3.692. PMID 12612263. 17) Общество синдрома Тернера США.« FAQ 6. Что можно сделать?». Проверено 11 мая 2007. 18) Давенпорт М.Л., Кроу Б.Дж., Трэверс С.Х., Рубин К., Росс Дж.Л., Фехнер П.Й., Гюнтер Д.Ф., Лю С., Геффнер М.Э., Траилкилл К., Хусеман К., Загар А.Дж., Куигли, Калифорния (2007). «Лечение гормоном роста ранней задержки роста у детей ясельного возраста с синдромом Тернера: рандомизированное контролируемое многоцентровое исследование». J Clin Endocrinol Metab. 92 (9): 3406–16. DOI: 10.1210 / jc.2006-2874. PMID 17595258. 19) Урбах А., Бенвенисти Н.; Бенвенисти (2009).«Изучение ранней летальности 45 эмбрионов XO (синдром Тернера) с использованием эмбриональных стволовых клеток человека». PLoS ONE 4 (1): e4175. DOI: 10.1371 / journal.pone.0004175. PMC 2613558. PMID 19137066. 20) Тернер HH (1938). «Синдром инфантилизма, врожденная перепонка шеи и вальгусный кубит». Эндокринология 23 (5): 566–74. DOI: 10,1210 / эндо-23-5-566.
синдром_тернера.txt · Последние изменения: 06.08.2019 14:21 — наталы
.ТЕРНЕРА СИНДРОМ — Большая Медицинская Энциклопедия
Тернера синдром (HH Turner, американский эндокринолог, род. В 1892 г .; синдром; син .: Шерешевского — синдром Тернера, синдром Улльриха, синдром Бонневи — Улльриковха, сексогенная карликовха, 9000 карликовха, сексогенная карликовха, др.) — генетически обусловленная форма первичного агонадизма (агенезии или дисгенезии гонад), относится к хромосомным болезням, сопровождается характерными аномалиями соматического и низкорослостыо.
Впервые эта наследственная болезнь (см.Наследственные болезни) была описана в 1925 г. Н. А. Шерешевским, к-рый считал, что она обусловлена недоразвитием половых желез (см.) И передней доли гипофиза (см.) И сочетается с врожденными пороками соматического развития. В 1938 г. Тернер подробно описал 7 больных и выделил характерную для этого симптомокомплекса триаду симптомов: половой инфантилизм (см.), Кожные крыловидные складки на боковых поверхностях шеи и деформацию локтевых суставов (вальгусный кубит). В СССР описываемый синдром принят называть синдромом Шерешевского — Тернера.
Частота встречаемости Тернера синдрома новорожденных составляет в среднем 0,03% (среди больных олигофренией частота встречаемости Тернера синдрома вдвое выше — 0,06%)
Этиология и патогенез
В 50—60-х гг. Фордом (С. Э. Форд) и др. у обследованных ими больных с Термин синдромом была обнаружена гоносомная моносомия 45, X, т. е. отсутствие одной из половых хромосом — X или Y (см. Хромосомный набор). Позднее были выявлены варианты Т. с. со структурными дефектами X-хромосомы (делеция короткого плеча, изо-Х-хромосома по длинному плечу, кольцевая X-хромосома и др.)) и с хромосомным мозаицизмом 45, Х / 46, XX; 45, Х / 46, XY; 45, Х / 46, ХХ / 47, ХХХ и др. (см. Мозаицизм). Соответственно кариотипу (см.) У большинства больных с Т. с. половой хроматин (см.) отсутствует, при мозаичных вариантах Т. с. с наличием в части клеток нормального женского кариотипа 46, XX содержание полового хроматина снижено, при структурных дефектах Х-хромосомы размеры Х-хроматина (телец Барра) могут быть увеличены или уменьшены.
Четкой связи возникновения Т. с. с возрастом и какими-либо заболеваниями родителей не выявлено.Однако беременности, заканчивающимся рождением детей с Т. с., нередко бывают осложнениями токсикозами беременных (см.), угрозой выкидыша. Роды (см.) Часто бывают преждевременными и патологическими. Особенности беременностей и родов, заканчивающихся рождением ребенка с Т. с., вероятно, следствие хромосомной патологии плода.
Нарушение формирования половых желез при Т. с. Условлено отсутствием или структурными дефектами одной половой хромосомы. Установлено, что в таком случае у эмбриона первичные герминативные клетки закладываются почти в нормальном количестве, однако во втором триместре происходит их быстрая инволюция и к моменту рождения ребенка количество примордиальных фолликулов в яичнике по сравнению с нормой увеличено или они полностью отсутствуют.Это приводит к выраженной эстрогенной недостаточности, половому недоразвитию, у многих больных — к первичной аменорее (см.) И бесплодию (см.).
Хромосомный дисбаланс в результате отсутствия одной из половых хромосом или части X-хромосомы является причиной возникновения различных дефектов соматического развития. Возможно также, что сопутствующие аутосомные мутации играют определенную роль в появлении пороков развития, соматически сходные с Т. с., но без видимой хромосомной патологии и без полового недоразвития (синдром Нунен, случаи Т.с. у мужчин),
Отсутствие Х-хромосомы приводит к проявлению у ряда больных с Т. с. рецессивных генов, использования в Х-хромосоме, что является причиной дальтонизма (см. Цветовое зрение) у больных (мужчин и женщин), недостаточностью глюкозо-6-фосфатдегидрогеназы (КФ 1.1.1.49) и др. и определите соответствующие мутантные генов в популяции.
Патологическая анатомия
При Тернера синдроме гонады обычно представляет собой недифференцированные соединительнотканные тяжи, не элементы гонады.Реже встречаются рудименты яичников (см.), Включающие овариальную строму, а иногда и отдельные примордиальные фолликулы. Последние могут наблюдаться у больных с кариотипом 45, X / 46, XX; 45, Х / 47, ХХХ. При кариотипе 45, X / 46, XY в гонадных тяжах могут быть обнаружены элементы яичек (см. Яичко) — канальцы, клетки Лейдига (интерстициальные эндокриноциты) и Сертоли (поддерживающие клетки) и рудименты семявыносящего протока (см.).
Прочие патологоанатомические данные соответствуют особенностям клин, проявлений.Наиболее важны изменения костио-суставной системы — конкресценция позвонков (см. Позвоночник), позвоночник (см.), Деформация суставов (см.), Укорочение метакарпальных и метатарзальных костей, фаланг (см. Кисть, Стопа), остеопороз (см.) и др., изменения сердца (см.) и крупных сосудов — коарктация аорты (см.), незаращение боталлова протока (см. Артериальный проток), межжелудочковой перегородки, стеноз устья аорты, почечных артерий и др., пороки развития почек (см. ) — подковообразная почка, удвоение лоханок и мочеточников и др.
Клиническая картина
Отставание больных с Тернера синдромом в физическом развитии заметно уже с рождения. Примерно у 15—20% больных с Т. с. задержка развития период наблюдается в пубертатном периоде. Для доношенных новорожденных характерны малая длина (42–48 см) и масса тела (2800–2500 г и менее), т. е. задержка физического развития носит внутриутробный характер. Это послужило причиной причисления Т. с. к примордиальному нанизму (см. Карликовость). Патогномоничными для Т. с.при рождении избыток кожи на шее (птеригиум — синдром) и другие пороки соматического развития, особенно костно-суставной и сердечно-сосудистой систем, «лицо сфинкса», лимфостаз. Для течения постнатального периода характерно беспокойство новорожденных, нарушение сосательного рефлекса, срыгивания фонтаном, рвота. В раннем возрасте у части больных отмечают задержку статического и речевого развития, что свидетельствует о патологии развития нервной системы в эмбриогенезе.
Рис.1. Внешний вид больной с синдромом Тернера: низко рослость, укороченная шея с характерными кожными складками, бочкообразная грудная клетка. Рис. 2. Больная с синдромом Тернера: виден характерный низкий рост волос на шее.Наиболее общим соматическим признаком при Т. с. является низко-рослость. Спонтанный рост больных, как правило, не выше 135— 145 см. Масса тела часто избыточна. Соматические симптомы у больных с Т. с. в порядке убывания по частоте встречаемости следующих: низкорослость (98%), общая диспластичность (92%), бочкообразная грудная клетка (75%), укорочение шеи (63%), низкий рост волос на шее (57%), высокое, «готическое» »Нёбо (56%), крыловидные складки кожи в области шеи (46%), деформация ушных раковин (46%), укорочение метакарпальных и метатарзальных костей и аплазия фаланг (46%), деформация ногтей (37%), вальгусная деформация локтевых суставов (36%), множественные пигментные родинки (35%), микрогнатизм (27%), лимфостаз (24%), птоз (24%), эпикантус (23%), пороки сердца и крупных сосудов (22%), артериальная гипертензия ( 17%), витилиго (8%).Типичный внешний вид больных с Т. с. представлен на рис. 1 и 2.
Рис. 3. Рентгенограммы кисти руки (прямая проекция) в норме (а) и больной с синдромом Тернера (б): на рентгенограмме кисти больной видны укорочения IV и V пястных костей, ногтевой фаланги I пальца.Рентгенологически при Т. с. турецкое седло и кости свода черепа обычно используется в отдельных случаях турецкое седло может иметь своеобразную форму с обызвествлением диафрагмы, быть уменьшенным или увеличенным. На рентгенограммах кистей и стоп встречается аплазия фаланг, гипоплазия метакарпальных и метатарзальных костей, деформация лучезапястного сустава типа Маделунга (см.Маделунга болезнь), на рентгенограммахочочника и грудной клетки — синостозы позвонков и ребер, явления гипертрофического остеопороза. Дифференцировка скелета умеренно отстает от возрастных норм, однако к пубертатному периоду она прогрессирует и обычно соответствует фактическому возрасту (см. Возраст костный). Характерная для Т. с. рентгенограмма представлена на рис. 3.
Половое недоразвитие при Тернера значительно отличается определенным своеобразием. Нередкими признаками являются геродермия (см.) и мошонкообразный вид больших половых губ, высокая промежность, недоразвитие малых половых губ, девственная плевы и клитора, воронкообразный вход во влагалище. В то же время у части имеются признаки маскулинизации в виде гипертрофии клитора, что часто сочетается с вирильным оволосением на теле. Молочные железы у больных с Т. с. не развиты, соски низко расположены, широко расставлены, бледны и втянуты (см. Молочная железа). Развивающиеся молочные железы при Т. с. имеют жировое строение и неправильную форму.Вторичное оволосение появляется спонтанно и бывает скудным. Матка недоразвита. Половые железы не развиты и представлены обычно соединительнотканными тяжами.
При Т. с. у мужчин кариотип обычно нормальный — 46, XY; мозаицизм 45, X / 46, XY встречается редко. Соматические проявления Т. с. у мужчин такие же, как у женщин, однако проявления гипогонадизма (см.) обычно отсутствуют.
В отличие от гипофизарного нанизма при Т. с. наблюдают высокий уровень гипоталамо-гипофизарной активности (см.Гипоталамо-гипофизарная система), что выражается в явлениях вегетососудистой дистонии (см. Ортостатические изменения кровообращения) по типу синдрома кастрации (см. Постстрационный синдром), в склонности к повышению АД у лиц молодого возраста и к ожирению (см.) С нарушениями трофики тканей , в характерных изменениях ЭЭГ. О повышенной гипофизарной активности при Т. с. показывают превышающие норму концентрации фолликулостимулирующего гормона (см.), лютеинизирующего гормона (см.), соматотропного гормона (см.), повышение функциональных резервов адренокортико-тропного гормона (см.), часто повышенная тиреотропная активность (см. Тропные гормоны гипофиза).
При Т. с. имеются заметные дерматоглифики (см.): чаще, чем в норме, встречается поперечная ладонная складка, трирадиус t (V) расположен дистально, угол при увеличении, резко увеличен общий гребневой счет.
Интеллект зараженных с Тернера синдром практически сохранен, имеющаяся иногда недостаточная недостаточность выражена нерезко, однако частота олигофрении (см.) при Т. с. все же выше, чем вообще в популяции. В психическом статусе больных с Т. с. основная играет роль своеобразный психический инфантилизм с эйфорией при хорошей практической приспособляемости и социальной адаптации.
Диагноз. Т. с. основывается на характерных клин. особенностями, определения полового хроматина и исследования кариотипа. Важность цитогенетических данных в случаях, не ясных клинически, особенно велика. В плане дифференциальной диагностики с карликовостью информативны данные определения содержания в крови тройных гормонов гипофиза, особенно гонадотропинов, и ЭЭГ, изменения к-рых при Т.с. и карликовости различны.
Лечение больных с Т. с. на первом этапе заключается в стимуляции роста тела анаболическими стероидами (см.) и другими анаболическими препаратами. Учитывая большую склонность этих больных к андрогенизации, лечение следует проводить минимально эффективными дозами анаболических стероидов с перерывами при регулярном гинекологическом контроле. Главным видом терапии больших с Т. с. является эстрогенизацией, к-рую следует проводить с 14–16 лет, вначале непрерывно до появления первого менструальноподобного кровотечения, далее — курсами, имитируя нормальный менструальный цикл.Лечение приводит к феминизации телосложения, развитию женских вторичных половых признаков, улучшает трофику половых путей, вызывает индуцированные менструации, снижает повышенную активность гипоталамо-гипофизарной системы, предотвращает развитие гипоталамической патологии, протекающей по типу посткастрационного синдрома. Лечение следует проводить в течение всего детородного возраста больных.
При Т. с. у мужчин в случае гипогонадизма заместительная терапия мужскими половыми гормонами (см.Андрогены).
Прогноз для жизни при Тернера синдроме благоприятный, исключение составляют больные с тяжелым и врожденным пороками сердца и с ренальной гипертензией. Лечение эстрогенами делает больных с Т. с. способными к семейной жизни, абсолютное большинство из них остаются бесплодными.
Библиогр .: Давиденкова Е. Ф., Берлинская Д. К. и Тысячнюк С. Ф. Клинические синдромы при аномалиях половых хромосом, Л., 1973; Зарубина Н. А. Гипофизарный нанизм, М., 1975; Уилкинс Л. Диагностика и лечение эндокринных нарушений в детском и юношеском возрасте, пер. с англ., М., 1963; Шарец Ю. Д. Дерматоглифика в медицине, Вестн. АМН СССР, № 7, с. 61, 1973; Шерешевский Н. А. К вопросу о сочетании уродства с эндокринопатиями, Вестн. эндокрин., т. 1, № 4, с. 296, 1925 г .; Форд С. Э. а. о. Аномалия половых хромосом в случае дисгенезии гонад (синдром Тернера), Lancet, v. 1, p. 711, 1959 г .; Labhart A. Klinik der inneren Sekretion, B. u. а., 1978; Полани П. Э., Хантер В.Ф. а. Леннокс Б. Хромосомный пол при синдроме Тернера с коарктацией аорты, Ланцет, т. 2, с. 120, 1954 г .; Учебник эндокринологии, под ред. Р. Уильямс, Филадельфия, 1981; Тернер Х. Синдром инфантилизма, врожденная перепончатая шея и вальгусный кубит, Эндокринология, т. 23, с. 566, 19 38.
Х.А. Зарубина.