Мышечная дистрофия Беккера — Информационный проект о нервно-мышечных заболеваниях
ИсследованияНекоторые из передовых стратегий включают в себя: введение гена дистрофина; изменение способов интерпретации клетками генных инструкций дистрофина; исправление самого гена; управление другими белками организма для компенсации отсутствия/нехватки дистрофина; совершенствование стероидных препаратов, и использование стволовых клеток для восстановления поврежденных мышц1.
Другие исследования сосредоточены на лечении сердечной недостаточности, связанной с недостатком дистрофина.
Повреждения гена дистрофина приводят к мышечной дистрофии Дюшенна (МДД), таким же образом, как и при менее тяжелой форме — мышечной дистрофии Беккера (МДБ). Множество стратегий лечения, опробованных при МДД, применимы для мышечной дистрофии Беккера.
Чтобы узнать больше информации об исследованиях, посвященных МДБ, предлагаем ознакомиться с данными видеоматериалами: Доклинические исследования мышечной дистрофии на животных моделях и МДБ: От целей до клинических испытаний.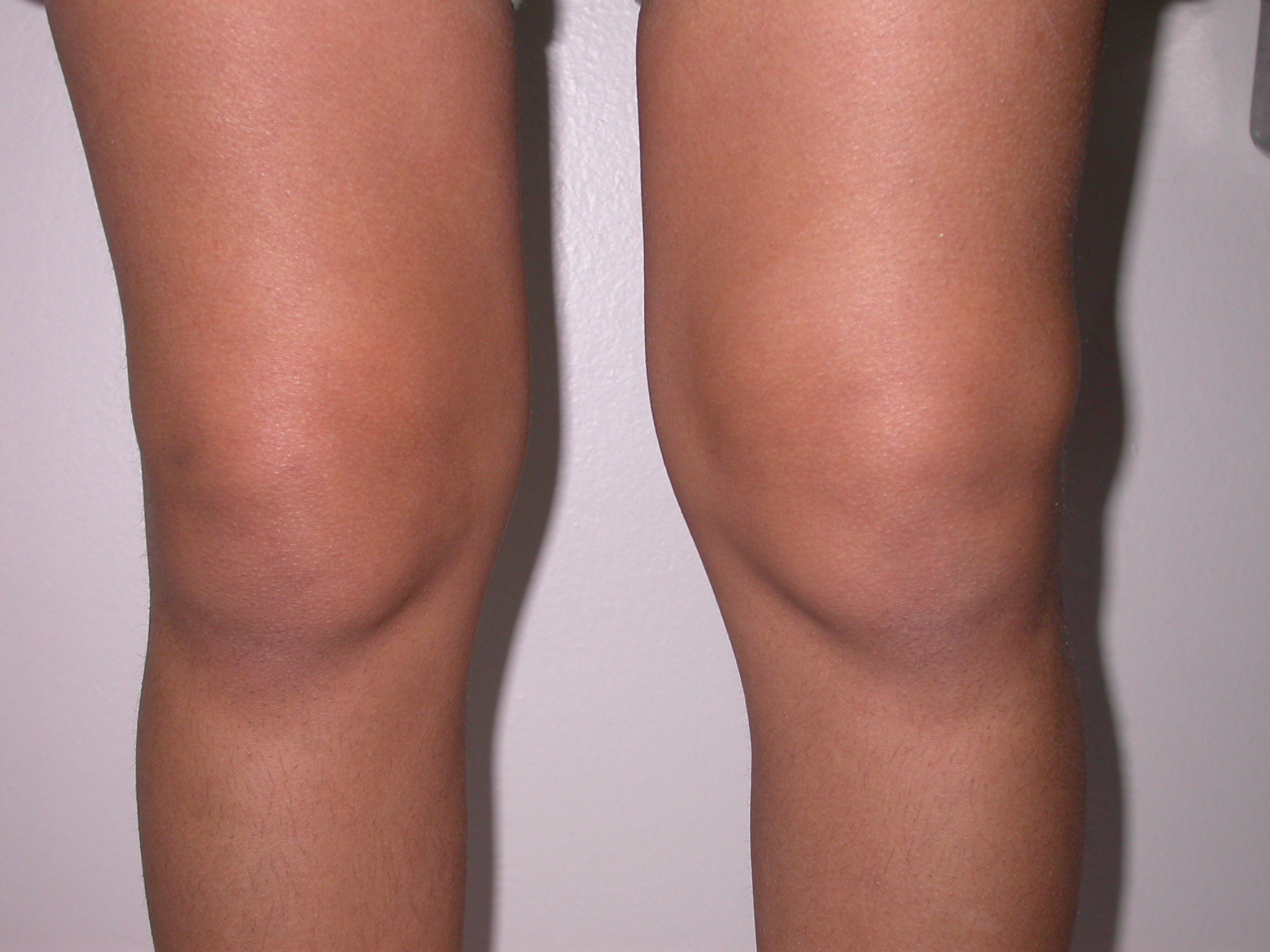
Исследователи преследуют несколько стратегий для поддержки и улучшения сердечной функции при МДБ и МДД. Они в основном тестируют существующие препараты на предмет возможной пользы для пораженного при МДД и МДБ сердца и проводят исследования с целью понять и найти новые подходы в лечении сердца при таких заболеваниях.
В 2009 году исследователи выяснили, что мутации в гене дистрофине, вызывающие кардимиопатию при МДБ, могут не поражать регионы белка, отвечающие за потерю скелетных мышц. Исследования могут позволить лучше прогнозировать появление кардиомиопатии при МДБ и применять кардиопротекторную терапию на более ранних этапах. Также они дают исследователям представление о том, какие части белка необходимо сохранить при рассмотрении укороченных молекул дистрофина в качестве терапевтических стратегий.
Обнаружено, что лекарственное средство силденафил (виагра) оказывает кардиопротекторный эффект на мышей с заболеванием, подобным МДД, как на ранней, так и на поздней стадии.
Лабораторные исследования показали, что экспериментальное соединение, предназначенное для герметизации клеточных мембран — р 188, улучшает работу сердца у собак с дефицитом дистрофина.
В 2011 году исследователи при поддержке MDA обнаружили, что ингибирование действия белка NF-kappa B улучшает сердечную функцию у мышей с тяжелой болезнью, подобной МДД. Фармацевтическая компания Catabasis, предоставляет на своем официальном сайте инфографический материал о препарате Edasalonexent (CAT-1004) — ингибиторе белка NF-kappa B.
Согласно информации, представленной в данном графике, в настоящее время изучается потенциал препарата Edasalonexent (CAT-1004) при лечении МДБ2.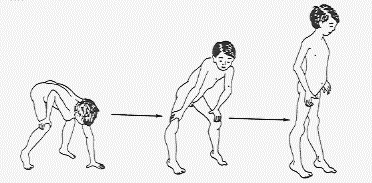
Метод CRISPR/Cas9 основан на естественной системе защиты бактерий от вирусной инфекции (аналог иммунной системы). Когда бактерия обнаруживает наличие чужеродной (в данном случае вирусной) ДНК, CRISPR белок захватывает часть вирусной ДНК и вставляет этот фрагмент в собственный геном бактерии. Затем бактерии используют «иммунизирующий» фрагмент вирусной ДНК для производства «антител», которые распознают и защищают от будущих вирусных атак3.
Бактериальные «антитела» включает в себя два типа коротких РНК,которые образуют комплекс с белком Cas9. Cas9 — это нуклеаза (тип фермента, способный разрезать ДНК). Когда соответствующая вирусу последовательность нуклеотидов (гидовая РНК) обнаруживает цель в геноме вируса, Cas9 вырезает вирусную ДНК, блокирует вирус и препятствует его репликации.
Как CRISPR/Cas9 может быть использован в лечении мышечной дистрофии?
Система CRISPR/Cas9 может быть задействована для изменения или исправления мутаций в клетках пациентов. Исследователи работают над тем, чтобы найти лучший способ лечения различных типов мышечной дистрофии (и других генетических заболеваний) с помощью данного метода.
Исследователи работают над тем, чтобы найти лучший способ лечения различных типов мышечной дистрофии (и других генетических заболеваний) с помощью данного метода.
Первые клинические испытания CRISPR/Cas9 с участием человека для лечения таких заболеваний как рак или муковисцидоз, находятся в процессе разработки. В данных испытаниях клетки пациента изолируются, обрабатываются для исправления генетической мутации, а затем вводятся обратно пациенту для борьбы с болезнью.
Для мышечной дистрофии вирусная система доставки могла бы обеспечить клетки пациента инструкциями для синтеза белка Cas9, так же как и гидовые РНК, которые нацелены на определенные области ДНК.
Рассмотрим пример с мышами, болевшими МДД. Заболевание обусловлено мутациями в гене, который содержит инструкции для производства дистрофина. После проведения лечения системой CRISPR/Cas9, удалось восстановить функцию дистрофина в мышечных клетках мышей.
Похожие доклинические исследования проводятся для оценки потенциала системы CRISPR/Cas9 в лечении других типов мышечной дистрофии.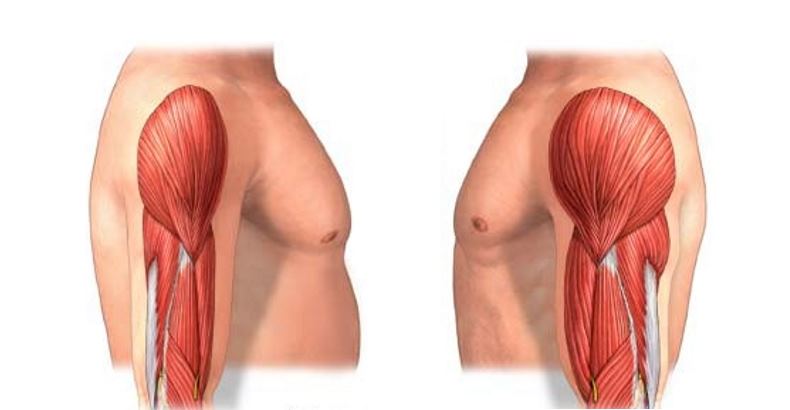
Для получения подробной информации — к ознакомлению доступны более 200 статей по CRISPR/cas-9 на сайте журнала Биомолекула https://biomolecula.ru/search/crispr
Генная терапияГенная терапия, или перенос генов, относится к доставке генов в качестве терапевтических агентов. Поскольку гены содержат инструкции по синтезу белка, это прямо или косвенно будет считаться терапией для нервно-мышечных заболеваний. Так как, перенесенные гены потенциально могут продолжать продуцировать белок в течение некоторого времени, генная терапия может предложить более надежное решение, чем другие методы лечения. При этом, генная терапия сталкивается со многими техническими проблемами, а также с особым контролем, установленным регулирующими органами, такими как Управление по контролю за продуктами и лекарствами США (FDA), что не позволяет генной терапии быть клинически применимым методом в настоящее время.
Ключевыми проблемами являются доставка генов в целевую ткань, избегая при этом нежелательных тканей и нежелательного иммунного ответа на белки, полученные из новых генов, или на средства доставки новых генов.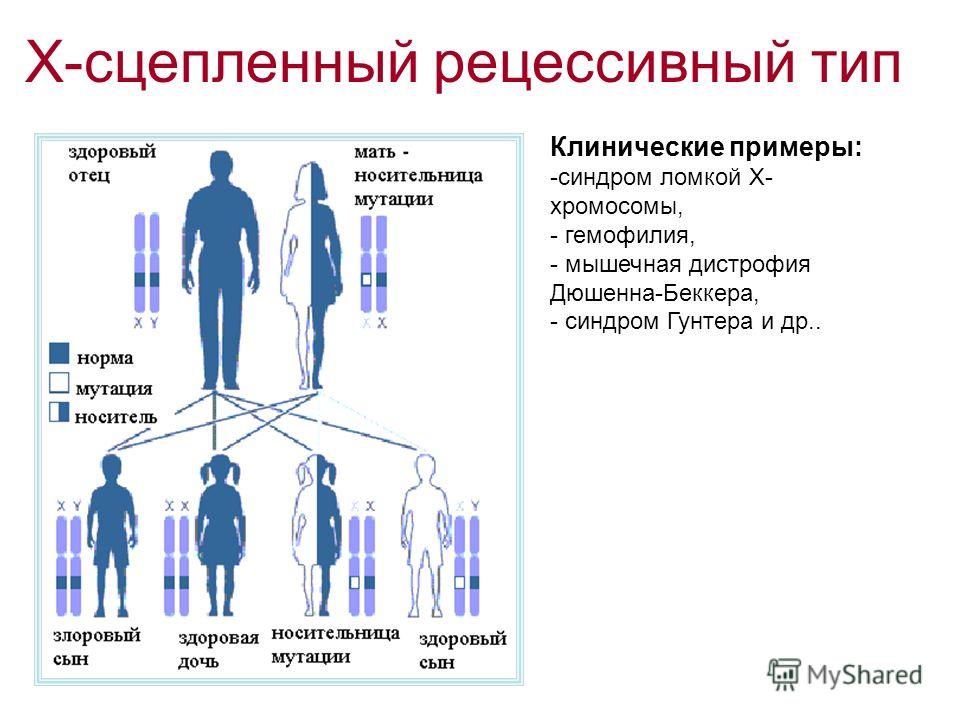
Ученые при поддержке MDA создали уменьшенный рабочий ген дистрофина, который был протестирован у мальчиков с МДД. Хотя лечение казалось безопасным, некоторые из мальчиков испытывали нежелательный иммунный ответ на белок дистрофина, который ограничивал эффективность переноса гена. Этот иммунный ответ подвергается дальнейшему исследованию.
Блокирование белка миостатина с помощью белка фоллистатина, представляет собой стратегию, которая имеет потенциал для лечения МДД и, вероятно, многих других нервно-мышечных заболеваний. Мыши с похожим на МДД заболеванием, получавшие гены белка фоллистатина, показали общее увеличение массы тела и веса отдельных мышц. У обезьян, которым был выполнен перенос гена фоллистатина, были более сильные и крупные мышцы.
Стратегия, получившая значительную поддержку MDA, заключается в подавлении действия белка природного происхождения, ограничивающего рост мышц- миостатина. Исследователи надеются, что блокирование миостатина может позволить мышцам расти больше и сильнее.
Ингибиторы миостатина привлекают большое внимание сообщества исследователей нервно-мышечных заболеваний с тех пор, как несколько лет назад было обнаружено, что люди и животные с генетическим дефицитом миостатина имеют большие мышцы и хорошую силу без видимых побочных эффектов. В 2010 году исследование показало, что мыши, лишенные дистрофина и имеющие заболевание, подобное МДД, получили пользу от лечения «приманкой», которая «выманивала» миостатин из их мышц.
Затем биотехнологическая компания Acceleron Pharma разработала препарат на основе этой «приманки» и начала его тестирование при поддержке MDA у мальчиков с МДД. К сожалению, во время этого испытания возникли неожиданные проблемы безопасности, в результате чего Acceleron прекратил его в 2011 году.
Компания надеется решить эти проблемы безопасности и возобновить тестирование ACE-031 или модифицированной версии ACE-031.
На сайте Clinicaltrials.gov можно ознакомиться с результатами исследования препарата ACE — 031 для пациентов с МДД4.
Другие стратегии ингибирования миостатина, такие как инъекция генов для миостатин-блокирующего фоллистатина, также находятся на рассмотрении.
Стволовые клеткиСтволовые клетки — это клетки на самых ранних стадиях развития. Они могут превратиться в специальный тип клеток (например, мышечные или нервные клетки), или сохранять плюрипотентность — способность развиваться в любой из ряда различных типов клеток.
Трансплантация стволовых клеток предлагается в качестве лечения таких заболеваний, как мышечная дистрофия. На основе клеточной терапии предпринимались попытки стимулировать регенерацию мышц с надеждой на то, что стволовые клетки восстановят мышечную функцию и исправят патологию путем повторного синтеза мышц. Стволовые клетки рассматриваются как подходящий вариант для терапевтического применения из-за их способности к самовосстановлению и потенциала дифференцировки.
В 2006 году при поддержке исследователи при поддержке Ассоциации мышечных дистрофий (MDA) восстановили подвижность у двух собак и стабилизировали функцию у третьей, используя стволовые клетки, взятые из мышечных кровеносных сосудов.
В исследовании, опубликованном в 2007 году, европейская исследовательская группа успешно использовала комбинацию генетической коррекции и стволовых клеток для лечения мышей с МДД. Исследователи в этом исследовании извлекали генерирующие мышцу стволовые клетки из мышечной ткани и крови у людей с МДД, исправляли генетическую ошибку в генах дистрофина клеток, а затем вводили клетки мышам с дефицитом дистрофина. Клетки, полученные из мышц, вызывали лучшую регенерацию мышц, чем клетки, полученные из крови.
В 2010 году французские ученые при поддержке MDA сообщали, что они идентифицировали ранее неизвестный тип мышечных стволовых клеток, расположенных в промежутках между мышечными волокнами у мышей. Хотя все это еще на ранних стадиях исследований, есть надежда, что новые клетки, получившие название PICs, могут играть важную роль в регенерации и восстановлении мышц.
В этом же году по утверждениям исследователей считалось, что для формирования новой мышечной ткани сначала требуется контролируемый тип повреждения ДНК. Новое открытие расширило понимание ученых о том, как незрелые мышечные клетки становятся мышцами, и помогло им управлять этим процессом для лечения нескольких форм мышечной дистрофии.
Новое открытие расширило понимание ученых о том, как незрелые мышечные клетки становятся мышцами, и помогло им управлять этим процессом для лечения нескольких форм мышечной дистрофии.
Стволовые клетки продолжают оставаться основной областью исследований для специалистов и исследователей в сфере нервно-мышечных заболеваний. Некоторые продолжают изучать мышечные сателлитные клетки, тип стволовых клеток, присутствующих в мышечной ткани. Другие изучают различные типы клеток, которые способны пережить трансплантацию в мышцы и продуцировать желаемые белки. Кроме того изучаются сходства и различия в развитии скелетных мышц и жировой ткани.
В последние годы (с 2013 по 2020 гг) были получены обнадеживающие результаты лечения человека с использованием зрелых стволовых клеток. Так проф. Sharma при участии соавторов., в 2013 году изучали эффект внутримышечной аутотрансплантации мезенхимальных стволовых клеток костного мозга у 150 пациентов с мышечной дистрофией (имеются ввиду стволовые клетки.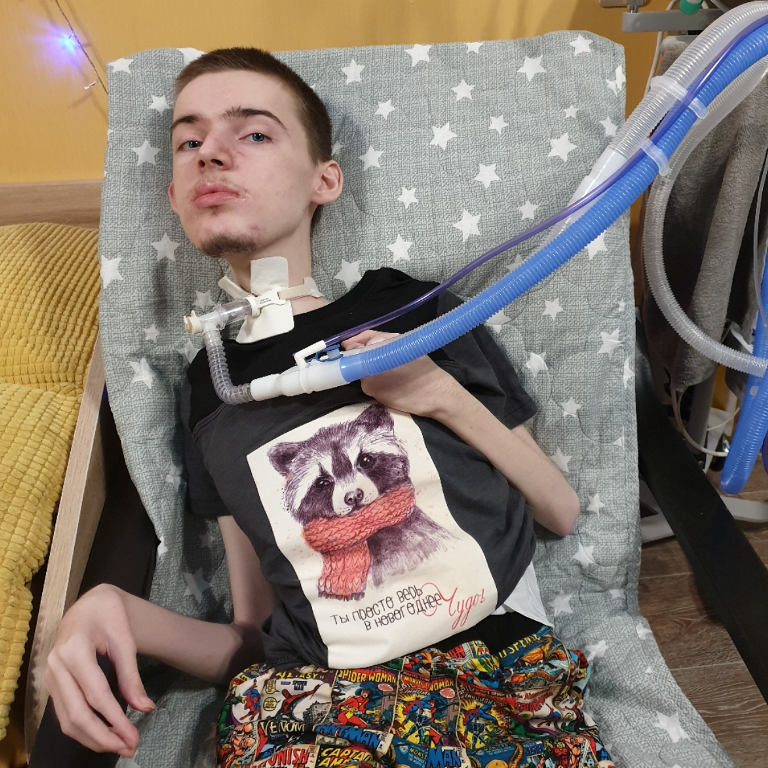 Через 12 месяцев наблюдения у пациентов наблюдалось увеличение мышечной силы и улучшение походки. Симптоматические и функциональные улучшения также наблюдались в 86,67% случаев: у шести пациентов снижен уровень жировой инфильтрации и выявлена регенерация мышц на снимках МРТ , а у девяти — выявлены положительные изменения электрической активности мышц на электронейромиографии (ЭНМГ).
Через 12 месяцев наблюдения у пациентов наблюдалось увеличение мышечной силы и улучшение походки. Симптоматические и функциональные улучшения также наблюдались в 86,67% случаев: у шести пациентов снижен уровень жировой инфильтрации и выявлена регенерация мышц на снимках МРТ , а у девяти — выявлены положительные изменения электрической активности мышц на электронейромиографии (ЭНМГ).
Мезанхимальные стволовые клетки состоят из множества клеток, таких как гематопоэтические стволовые клетки, тканеспецифические клетки-предшественники, стромальные клетки и специализированные клетки крови на разных стадиях развития5. Эти клетки обладают способностью мобилизовать и оказывать свои репаративные эффекты в месте повреждения. Они способствуют неоваскуляризации и усиливают ангиогенез (образование сосудов), продуцируя сигнальные молекулы, такие как факторы роста эндотелия сосудов и факторы роста фибробластов (FGF2). Они также способствуют ремоделированию тканей, предотвращают апоптоз (отмирание клеток), уменьшают воспаление, высвобождают факторы роста и активируют сателлитные клетки. Это паракринные эффекты, которые могут помочь в достижении желаемого результата клеточной терапии6,7. Аутологичные мезенхимальные стволовые клетки костного мозга были использованы в этом случае, потому что они не имеют этических проблем, и его безопасность была установлена (не требуется донор, это клетки самого пациента).
Это паракринные эффекты, которые могут помочь в достижении желаемого результата клеточной терапии6,7. Аутологичные мезенхимальные стволовые клетки костного мозга были использованы в этом случае, потому что они не имеют этических проблем, и его безопасность была установлена (не требуется донор, это клетки самого пациента).
Трансплантация стволовых клеток в нужное место мышечного тела, как правило, является основной практической трудностью. Внутривенное введение стволовых клеток, полученных из костного мозга, показало успешное возвращение стволовых клеток в поврежденные мышечные ткани на моделях животных; однако это также рискует разбавлением концентрации клеток. Мышечная дистрофия в основном воспринимается как заболевание мышц, с малым количеством свидетельств нервно-мышечных поражений. Дистрофин является частью структурного белка, обнаруженного в миелине, образующего клетки Шванна и нервы. Демиелинизация и дегенерация как изменения в нервах могут происходить с такими нарушениями в клетках. Поэтому были выбраны два различных способа трансплантации клеток: внутримышечный и интратекальный. Мезенхимальные клетки костного мозга вводили в двигательные точки целевых слабых мышц для восстановления иннервирующего нерва, а также мышц. Известно, что спинномозговая жидкость содержит факторы роста, которые помогают росту коркового эпителия и стимулируют васкуляризацию в нервной системе, поэтому он использовался в качестве разбавляющей среды.
Поэтому были выбраны два различных способа трансплантации клеток: внутримышечный и интратекальный. Мезенхимальные клетки костного мозга вводили в двигательные точки целевых слабых мышц для восстановления иннервирующего нерва, а также мышц. Известно, что спинномозговая жидкость содержит факторы роста, которые помогают росту коркового эпителия и стимулируют васкуляризацию в нервной системе, поэтому он использовался в качестве разбавляющей среды.
Особый интерес вызывает клинический случай пациента с мышечной дистрофией Беккера, в отношении которого была проведена терапия с использованием мезанхемальных клеток костного мозга.
Результаты были обнадеживающими. После клеточной терапии пациент дважды наблюдался в стационаре через 3 и 9 месяцев. Спустя 3 месяца было отмечено улучшение двигательной функции верхних конечностей. Выполнение движений над головой требовало сравнительно меньших усилий. Наблюдалось двустороннее снижение жесткости и псевдогипертрофии икроножных мышц . Отмечались значительные улучшения в положении стоя и сидя, в способности держать равновесие. Баланс в положении стоя и при ходьбе улучшился. Частота падений при ходьбе заметно уменьшилась с 4-5 падений в месяц до 1 падения за 3 месяца. Характеристики дыхательной функции также улучшились: жизненная емкость легких (ЖЕЛ) (с 1250 мл до 1750 мл) и пиковая скорость выдоха (ПСВ) (с 290 мл до 360 мл).
Баланс в положении стоя и при ходьбе улучшился. Частота падений при ходьбе заметно уменьшилась с 4-5 падений в месяц до 1 падения за 3 месяца. Характеристики дыхательной функции также улучшились: жизненная емкость легких (ЖЕЛ) (с 1250 мл до 1750 мл) и пиковая скорость выдоха (ПСВ) (с 290 мл до 360 мл).
Более подробно читайте запись «Эффективность клеточной терапии при прогрессирующей мышечной дистрофии Беккера»8.
Усиление атрофинаЛабораторные данные показывают, что повышение уровня мышечного белка атрофина может, до некоторой степени, компенсировать дефицит дистрофина.
Утрофин очень похож на дистрофин, но, в отличие от дистрофина, обычно вырабатывается и полностью функционирует при МДБ. Следовательно, повышение уровня атрофина вряд ли вызовет нежелательный иммунный ответ, тогда как повышение уровня дистрофина может это сделать. Увеличение производства атрофина может помочь компенсировать дефицит дистрофина независимо от специфической мутации гена дистрофина.
Хотя по структуре и функции атрофин близок к дистрофину, между этими двумя белками есть как минимум одно ключевое отличие. Во время развития плода и, возможно, немного позднее, атрофин присутствует по всему мышечному волокну, взаимодействуя с кластерами белков, застрявшими в окружающей его мембране. По мере взросления животного или человека атрофин почти полностью заменяется дистрофином, за одним исключением. На нервно-мышечном соединении атрофин остается на протяжении всей жизни.
Несколько стратегий в настоящее время пытаются увеличить атрофин. Одним из них является выявление и подавление всего, что препятствует выработке атрофина — найти тормоз и, так сказать, отпустить его.
Другая стратегия заключается в том, чтобы ввести модифицированную версию самого белка атрофина в организм. Исследование, проведенное в 2009 году, показало, что модифицированный протеин атрофина дает значительные преимущества при введении мышам, у которых отсутствует белок дистрофина и которые имеют заболевание, напоминающее МДД.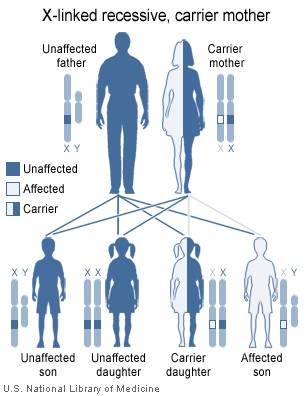
В 2011 году ученые сообщили, что системное введение человеческой формы белка, называемого бигликан, мышам с болезнью, подобной МДД , повышает устойчивость мышечных мышц к повреждениям, связанным с их сокращением.
ReveraGEN и VamoroloneПрепарат Vamorolone снижает воспаление в мышцах, что эффективно тормозит прогрессию мышечной дистрофии. При этом в отличие от традиционных глюкокортикостероидов, применяемых в лечении мышечных дистрофий, препарат не имеет таких нежелательных побочных эффектов.
В марте 2019 года от МОО «Проект Ай-Мио» на конференции Myology 2019 побывали Бережной Дмитрий и Екатерина Чернец. Нам стало известно, что компания Reveragen планирует провести исследование препарата Vamorolone для пациентов с мышечной дистрофией Беккера.
В мае 2019 года удалось выйти на связь с Eric’ом Hoffman’ом, руководителем компании Reveragen.
Мы получили следующую информацию:
«Мы (Reveragen) подали заявку на грант для возможного финансирования правительством США небольшого «пилотного испытания» Vamorolone при мышечной дистрофии Беккера.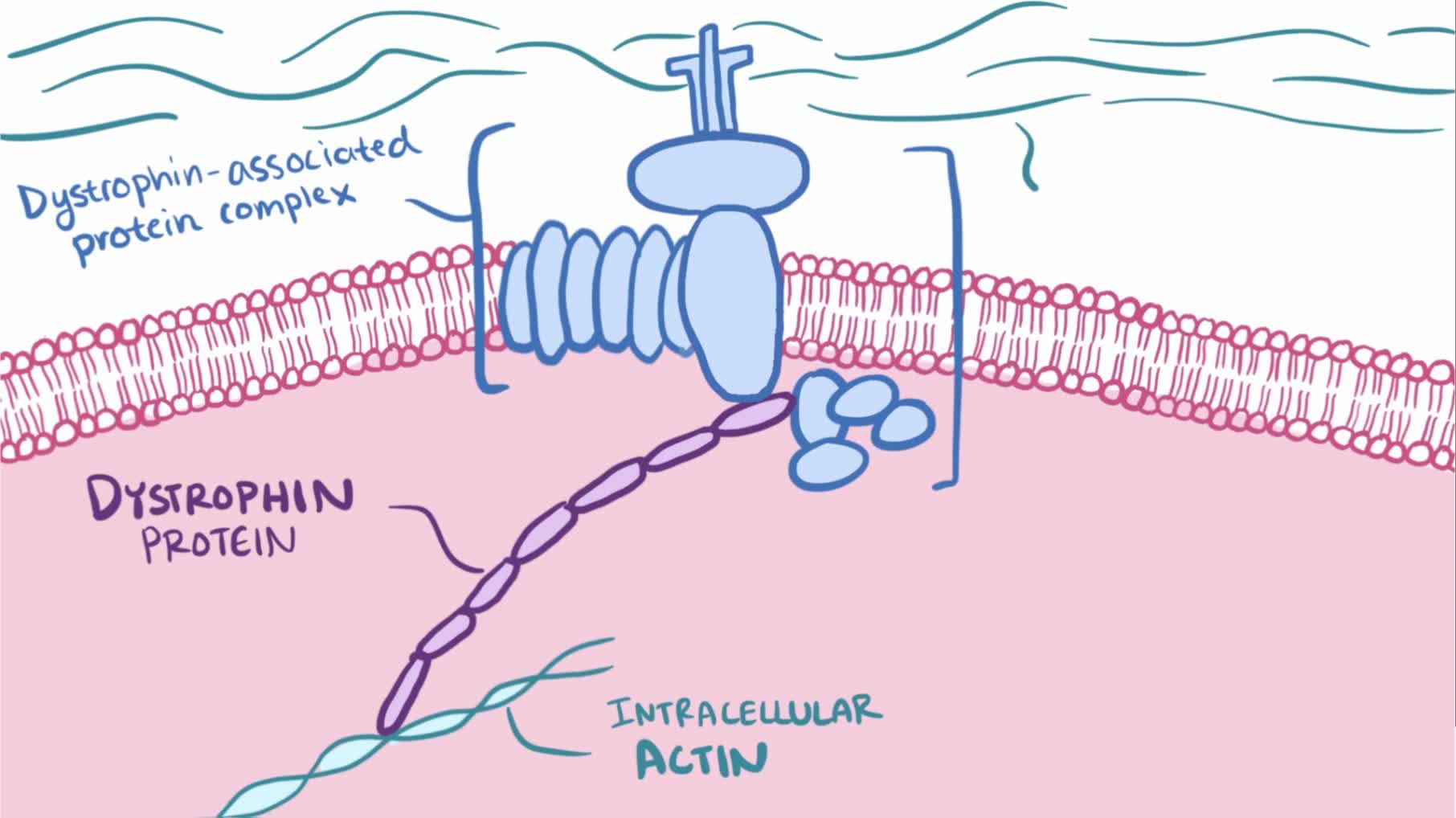 Нам нужно подождать несколько месяцев, чтобы узнать, предоставит ли правительство финансирование. Если это состоится, то экспериментальные испытания могут начаться через год или около того, и с ограниченным количеством пациентов в пилотном исследовании, набор пациентов может быть также ограничен Питтсбургом и Падуей, Италия — но все эти детали еще не проработаны.»
Нам нужно подождать несколько месяцев, чтобы узнать, предоставит ли правительство финансирование. Если это состоится, то экспериментальные испытания могут начаться через год или около того, и с ограниченным количеством пациентов в пилотном исследовании, набор пациентов может быть также ограничен Питтсбургом и Падуей, Италия — но все эти детали еще не проработаны.»
Мы будем следить за новостями, обновления будут размещены здесь и в разделе сайта «Блог».
Источники:
1. Muscular dystrophy Association — исследования терапевтических стратегий для мышечной дистрофии Беккера
2. Catabasis.com — график исследований CAT — 1004
3. Musculardystrophynews.com — о CRISPR/Cas-9
4. Clincaltrials.gov — результаты исследований ACE — 031
5. Sharma A., Gokulchnadran N, Sane H, Badhe P. 3rd eds Stem cell therapy in neurological disorders. NeuroGen Brain and Spine Institute, Navi Mumbai, India: 2015 [Google Scholar]
6. Vandervelde S, Luyn V. M., Tio J, Harmsen MC. Signaling factors in stem cell mediated repair of infarcted myocardium. J Mol Cell Cardiol 2005; 39:363-76. [PubMed] [Google Scholar]
Signaling factors in stem cell mediated repair of infarcted myocardium. J Mol Cell Cardiol 2005; 39:363-76. [PubMed] [Google Scholar]
7. Gnecchi M, Zhang Z, Ni A, Dzau VJ. Paracrine mechanisms in adult stem cell signaling and therapy. Circulation Research 2008;103:1204-19. [PMC free article] [PubMed] [Google Scholar]
8. Clinical trials.gov — эффективность клеточной терапии при мышечной дистрофии Беккера
Перевод и материалы подготовлены: Бережной Д.С., Чернец Е.Н.
Мышечная дистрофия Дюшенна-Беккера. Лайонизация Х-хромосомы у девочек (Duchenne Muscular Dystrophy, X-Lyonization, Girls)
Исследуемый материал Цельная кровь (с ЭДТА)
Метод определенияПЦР-ПДАФ, метилчувствительный рестрикционный анализ
Выдаётся заключение врача-генетика!
Тип наследования.
Х-сцепленный рецессивный, т.е. им страдают почти исключительно мальчики, женщины же с поврежденным геном в одной из Х-хромосом являются носительницами МДД. Но в редких случаях миодистрофией Дюшенна могут болеть и девочки. Причинами этого могут быть преимущественная инактивация Х-хромосомы с нормальным аллелем у гетерозиготных носительниц мутантного гена дистрофина, Х-аутосомная транслокация, затрагивающая этот ген, гемизиготность по мутантному аллелю и наличие фенокопий (заболеваний, связанных с нарушением других белков, входящих в дистрофин-гликопротеиновый комплекс). Приблизительно в 2/3 случаев сын получает хромосому с повреждением от матери-носительницы, в остальных случаях заболевание возникает в результате мутации de novo в половых клетках матери или отца, либо в предшественниках этих клеток. Приблизительно 30% всех случаев заболевания связаны с возникновением свежих мутаций в гене дистрофина, а остальные 70% обусловлены носительством матерью пробанда патологической мутации в одной из Х хромосом. Считается, что 6-7% всех спорадических случаев заболевания являются следствием гонадного мозаицизма — существования в яичниках женщины нескольких генераций ооцитов с нормальными и мутантными аллелями гена дистрофина.
Но в редких случаях миодистрофией Дюшенна могут болеть и девочки. Причинами этого могут быть преимущественная инактивация Х-хромосомы с нормальным аллелем у гетерозиготных носительниц мутантного гена дистрофина, Х-аутосомная транслокация, затрагивающая этот ген, гемизиготность по мутантному аллелю и наличие фенокопий (заболеваний, связанных с нарушением других белков, входящих в дистрофин-гликопротеиновый комплекс). Приблизительно в 2/3 случаев сын получает хромосому с повреждением от матери-носительницы, в остальных случаях заболевание возникает в результате мутации de novo в половых клетках матери или отца, либо в предшественниках этих клеток. Приблизительно 30% всех случаев заболевания связаны с возникновением свежих мутаций в гене дистрофина, а остальные 70% обусловлены носительством матерью пробанда патологической мутации в одной из Х хромосом. Считается, что 6-7% всех спорадических случаев заболевания являются следствием гонадного мозаицизма — существования в яичниках женщины нескольких генераций ооцитов с нормальными и мутантными аллелями гена дистрофина.
Гены, ответственные за развитие заболевания.
DMD (DYSTROPHIN) — ген дистрофина, находится в Х-хромосоме в регионе Хр21.2 –р21.1, состоит из 79 экзонов. У 60%-70% больных выявляются крупные делеции, захватывающие один или несколько экзонов гена и локализованные в двух «горячих» регионах — в области 5′ конца (экзоны 6-19) и 3′ конца (экзоны 40-43). У 5% больных обнаруживаются дупликации, в остальных случаях — точковые мутации. Различия в тяжести клинических проявлений при двух аллельных вариантах заболевания связывают с различиями в характере мутации в гене дистрофина. При мышечной дистрофии Дюшенна мутации в гене дистрофина приводят к сдвигу рамки считывания и преждевременной терминации трансляции, при этом синтез белка прекращается. При мышечной дистрофии Беккера структурные перестройки гена не приводят к сдвигу рамки считывания, ДНК-полимераза может «перескакивать» делетированные экзоны, что приводит к синтезу внутренне усеченного белка, который может, до некоторой степени, выполнять свои функции.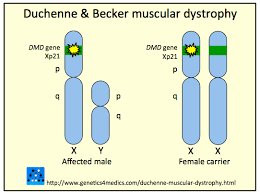
Определение заболевания.
Нейромышечное заболевание, обусловленное мутацией в гене дистрофина и приводящее к прогрессирующей дегенерации мышечных волокон.Патогенез и клиническая картина.
Основная функция дистрофина заключается в обеспечении устойчивости и эластичности мышечного волокна при последующих мышечных сокращениях. При отсутствии дистрофина вследствие мутации мембрана разрушается, в ней появляются участки некроза, что приводит к вымыванию содержимого саркоплазмы в кровяное русло. Происходит постепенная гибель мышечных волокон и замещение их соединительнотканными структурами, которые увеличивают плотность и объем мышц, вызывая феномен псевдогипертрофии. Заболевание встречается в двух клинических формах, являющихся аллельными генетическими вариантами.Прогрессирующая мышечная дистрофия Дюшенна.
Заболевание проявляется в возрасте 1-5 лет, быстро прогрессирует и приводит к летальному исходу до 25 летнего возраста. Для большинства больных характерна задержка темпов раннего моторного развития. При начале самостоятельной ходьбы, в возрасте старше 14 месяцев, отмечаются частые падения, спотыкания, моторная неловкость, быстрая утомляемость. Постепенно походка становится переваливающейся, возникают затруднения при подъеме по лестнице и из положения на корточках, когда больные вынуждены использовать вспомогательные приемы Говерса («взбирание по самому себе»). На ранних стадиях заболевания обнаруживаются псевдогипертрофии мышц, возникающие за счет разрастания соединительной и жировой ткани на месте гибнущих мышечных волокон. Наиболее часто они локализуются в икроножных, дельтовидных, четырехглавых и трехглавых мышцах и создают ложное впечатление атлетического телосложения больного. По мере прогрессирования заболевания псевдогипертрофии мышц трансформируются в их гипотрофии. Распространение патологического процесса имеет восходящий характер. Первыми поражаются мышцы тазового пояса и проксимальных отделов нижних конечностей, затем мышцы плечевого пояса, спины и проксимальных отделов верхних конечностей.
При начале самостоятельной ходьбы, в возрасте старше 14 месяцев, отмечаются частые падения, спотыкания, моторная неловкость, быстрая утомляемость. Постепенно походка становится переваливающейся, возникают затруднения при подъеме по лестнице и из положения на корточках, когда больные вынуждены использовать вспомогательные приемы Говерса («взбирание по самому себе»). На ранних стадиях заболевания обнаруживаются псевдогипертрофии мышц, возникающие за счет разрастания соединительной и жировой ткани на месте гибнущих мышечных волокон. Наиболее часто они локализуются в икроножных, дельтовидных, четырехглавых и трехглавых мышцах и создают ложное впечатление атлетического телосложения больного. По мере прогрессирования заболевания псевдогипертрофии мышц трансформируются в их гипотрофии. Распространение патологического процесса имеет восходящий характер. Первыми поражаются мышцы тазового пояса и проксимальных отделов нижних конечностей, затем мышцы плечевого пояса, спины и проксимальных отделов верхних конечностей.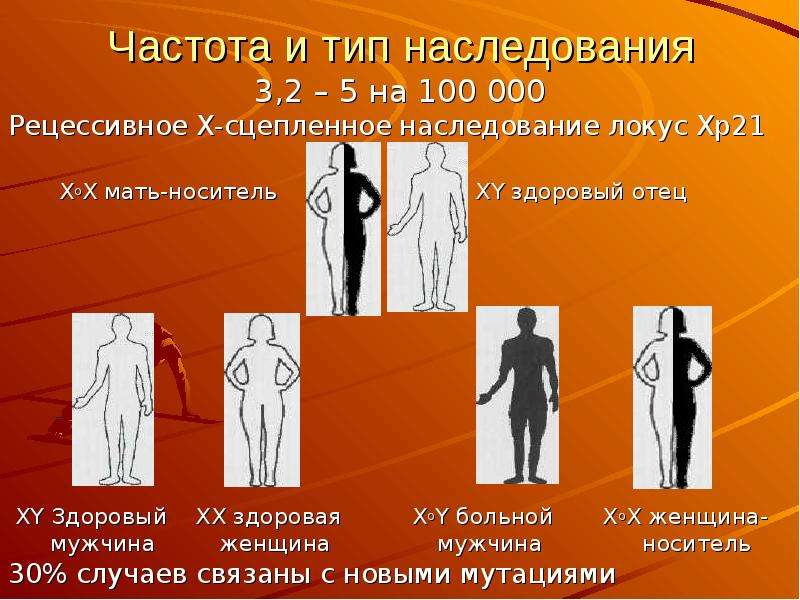 Уже на ранних стадиях болезни снижаются или угасают коленные рефлексы. Ахиллов рефлекс, а также сухожильные рефлексы с рук, могут длительное время оставаться сохранными. По мере развития патологического процесса в мышцах возникают вторичные деформации позвоночника (усиление лордоза и кифоза, сколиоз), грудной клетки (по типу седловидной и килевидной) и стоп, а также ретракции сухожилий с развитием контрактур в суставах. Характерным признаком является кардиомиопатия, которая проявляется симптомами гипертрофии левого желудочка и аритмией. У 25-30% больных диагностируется олигофрения в степени дебильности. Пациенты сохраняют способность к самостоятельной ходьбе до 10-12-ти летнего возраста, после чего пользуются инвалидной коляской. Гибель больных наступает от сердечной недостаточности или от интеркуррентных инфекций.
Уже на ранних стадиях болезни снижаются или угасают коленные рефлексы. Ахиллов рефлекс, а также сухожильные рефлексы с рук, могут длительное время оставаться сохранными. По мере развития патологического процесса в мышцах возникают вторичные деформации позвоночника (усиление лордоза и кифоза, сколиоз), грудной клетки (по типу седловидной и килевидной) и стоп, а также ретракции сухожилий с развитием контрактур в суставах. Характерным признаком является кардиомиопатия, которая проявляется симптомами гипертрофии левого желудочка и аритмией. У 25-30% больных диагностируется олигофрения в степени дебильности. Пациенты сохраняют способность к самостоятельной ходьбе до 10-12-ти летнего возраста, после чего пользуются инвалидной коляской. Гибель больных наступает от сердечной недостаточности или от интеркуррентных инфекций.Прогрессирующая мышечная дистрофия Беккера.
Наиболее часто заболевание возникает в возрастном интервале от 10 до 20 лет с появления слабости и утомляемости мышц тазового пояса и ног.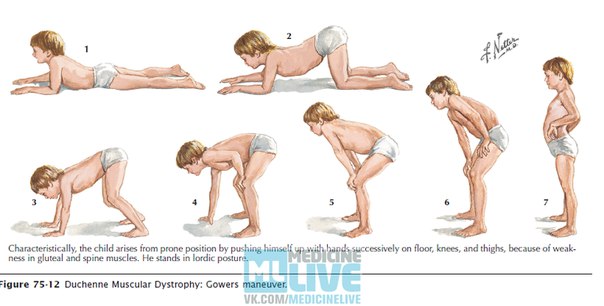 Ранними симптомами у значительного числа больных бывают болезненные мышечные крампи. Клинические проявления сходны с таковыми при ПМДД, однако имеют значительно меньшую степень выраженности. Характерной особенностью ПМДБ является вовлечение в патологический процесс миокарда. Гипертрофическая или дилятационная кардиомиопатия диагностируется у 50-60% больных. В 40-50% случаев выявляются гипогенитализм и атрофия яичек. Интеллект, как правило, не страдает. Заболевание прогрессирует достаточно медленно и в большинстве случаев приводит к инвалидизации больного не ранее 40-летнего возраста.
Ранними симптомами у значительного числа больных бывают болезненные мышечные крампи. Клинические проявления сходны с таковыми при ПМДД, однако имеют значительно меньшую степень выраженности. Характерной особенностью ПМДБ является вовлечение в патологический процесс миокарда. Гипертрофическая или дилятационная кардиомиопатия диагностируется у 50-60% больных. В 40-50% случаев выявляются гипогенитализм и атрофия яичек. Интеллект, как правило, не страдает. Заболевание прогрессирует достаточно медленно и в большинстве случаев приводит к инвалидизации больного не ранее 40-летнего возраста.Прогрессирующая мышечная дистрофия Дюшенна у лиц женского пола.
Описаны клинические проявления ПМДД у лиц женского пола, которые являются носительницами мутации в гене дистрофина в гетерозиготном состоянии. Клинические признаки могут появиться в различные возрастные периоды, но чаще провоцируются гормональными перестройками в организме женщины (начало менструаций, беременность, климакс). Появление клинических симптомов может быть обусловлено двумя причинами: 1) наличие полной или мозаичной форм синдрома Шерешевского-Тернера; 2) феноменом несбалансированной лайонизации. На электромиограмме выявляются признаки первично-мышечного поражения в виде усиления интерференции и снижения амплитуды М-ответа. Высокую диагностическую значимость имеет определение активности фермента креатинфосфокиназы в плазме крови больного. Этот показатель у больных ПМДД в 50-100 раз превышает норму и может быть выявлен до возникновения выраженных клинических признаков. Для диагностики и дифференциальной диагностики ПМДД/ПМДБ используются иммуногистохимические методы анализа дистрофина в биоптате мышечного волокна. При использовании антисывороток на различные районы дистрофина при ПМДД иммунореактивных форм белка, как правило, не выявляется. У больных с ПМДБ наблюдается прерывистое окрашивание мышц при иммунохимическом анализе, что свидетельствует об относительной сохранности отдельных структур цитоскелета. Специфического морфологического дефекта не существует. В биоптате мышц больных выявляются изменения, характерные для группы прогрессирующих мышечных дистрофий в целом.
На электромиограмме выявляются признаки первично-мышечного поражения в виде усиления интерференции и снижения амплитуды М-ответа. Высокую диагностическую значимость имеет определение активности фермента креатинфосфокиназы в плазме крови больного. Этот показатель у больных ПМДД в 50-100 раз превышает норму и может быть выявлен до возникновения выраженных клинических признаков. Для диагностики и дифференциальной диагностики ПМДД/ПМДБ используются иммуногистохимические методы анализа дистрофина в биоптате мышечного волокна. При использовании антисывороток на различные районы дистрофина при ПМДД иммунореактивных форм белка, как правило, не выявляется. У больных с ПМДБ наблюдается прерывистое окрашивание мышц при иммунохимическом анализе, что свидетельствует об относительной сохранности отдельных структур цитоскелета. Специфического морфологического дефекта не существует. В биоптате мышц больных выявляются изменения, характерные для группы прогрессирующих мышечных дистрофий в целом.
Частота встречаемости: Мышечная дистрофия Дюшенна (МДД): 1:2500-4000 новорожденных мальчиков. Частота МДБ (Беккера) составляет 1 на 20000 мальчиков.
Частота МДБ (Беккера) составляет 1 на 20000 мальчиков.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Monaco, A. P.; Kunkel, L. M.: A giant locus for the Duchenne and Becker muscular dystrophy gene. Trends Genet. 3: 33-37, 1987.
- Becker, P. E.: Eine neue X-chromosomale Muskeldystrophie. Acta Psychiat. Neurol. Scand. 193: 427, 1955.
Генодиагностика при мышечной дистрофии Дюшенна/Беккера
Миодистрофия Дюшенна/Беккера относится к X-сцепленным рецессивным наследственным заболеваниям и обусловлена мутацией в гене, кодирующем белок дистрофин (ген DMD). Тест позволяет выявлять мутации в гене дистрофина, которые ответственны за развитие наиболее частой причины поражения мышечной системы в молодом возрасте.
Синонимы русские
Дистрофинопатии, миодистрофия Дюшенна (МД), миодистрофия Беккера (МБ), ген DMD, генетическое обследование.
Синонимы английские
Dystrophinopathies, Duchenne muscular dystrophy, Becker muscular dystrophy, gene DMD.
Название гена
Ген DMD.
Локализация гена на хромосоме
Локус Xp21.2-p21.1.
Метод исследования
Полимеразная цепная реакция (ПЦР), фрагментный анализ гена DMD.
Какой биоматериал можно использовать для исследования?
Венозную кровь.
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Миодистрофия Дюшенна/Беккера относится к X-сцепленным рецессивным наследственным заболеваниям и обусловлена мутацией в гене, кодирующем белок дистрофин (ген DMD). Ген дистрофина состоит из 79 экзонов – это один из самых крупных генов человека, расположен на Х-хромосоме (локус Xp21.2). При мышечной дистрофии обнаруживаются мутации (чаще всего делеции) одного или нескольких экзонов гена, реже точковые мутации или дупликации. В данном исследовании анализируются мутации экзонов 1-10, 21-30, 41-50, 61-70.
Дистрофинопатии представляют собой спектр наследственных Х-сцепленных заболеваний, вызываемых различными патологическими аберрациями в гене DMD. У носителей мутации мужского пола риск развития заболевания близок к 100%, у носителей женского пола проявления заболевания более мягкие либо не наблюдаются совсем. Тяжесть проявлений дистрофинопатий зависит от типов мутаций и может варьироваться от асимптоматического повышения креатинфосфокиназы или мышечных судорог с миоглобинурией до развития классических синдромов, таких как мышечная дистрофия Беккера и Дюшенна.
Миодистрофия Дюшенна (МД) чаще всего манифестирует в раннем детстве до 5 лет с задержки достижения основных этапов моторного развития и характеризуется полным отсутствием синтеза функционально активного дистрофина. На начальных этапах МД в основном поражаются проксимальные отделы мышечной системы (мышцы бедра, таза, плечевого пояса), но при последующей прогрессии затрагиваются все отделы мышечной системы. Помимо выраженной миодистрофии, у пациентов с МД наблюдается повышение уровня креатинфосфокиназы, псевдогипертрофия мышц голеней, различные скелетные аномалии и кардиомиопатия, чаще всего возникающая после 18 лет. Примерно у 75% пациентов с МД наблюдается делеция или дупликация одного или нескольких экзонов гена DMD.
Миодистрофия Беккера (МБ) представляет собой более легкую форму дистрофинопатии, характеризующуюся достаточным синтезом функционально активного белка. При МБ наблюдается мышечная слабость проксимальных отделов мышц, низкая толерантность к нагрузкам, миоглобулинурия, миалгия и повышение уровня креатинфосфокиназы. Делеции и дупликации одного или нескольких экзонов гена DMD являются наиболее частыми наблюдаемыми при МБ генетическими аберрациями (70-80% всех случаев).
Дистрофинопатия может проявляться у 5-10% носителей мутации женского пола мышечной слабостью, миалгией, судорогами и дилатационной кардиомиопатией.
Дистрофинопатии – X-сцепленные заболевания и наследуется по аутосомно-доминантному типу, то есть имеется 50% риска наследования данного заболевания от матери с аберрантным геном.
Для чего используется исследование?
В соответствии с международными клиническими рекомендациями, генетическое обследование на миодистрофию Дюшенна/Беккера проводится при наличии у пациента клинической симптоматики, характерной для данного заболевания, а также родственникам и детям больного.
Когда назначается исследование?
- При подозрении на миодистрофию Дюшенна/Беккера;
- при дифференциальной диагностике мышечной слабости;
- при дифференциальной диагностике мышечных судорог и миоглобинурии;
- при раннем выявлении заболевания у родственников;
- при планировании семьи.
Что означают результаты?
Генетическое обследование является основным методом подтверждения диагноза и основано на выявлении делеции или дупликации одного или нескольких экзонов с помощью метода фрагментного анализа в гене DMD.
Референсные значения
Патологических делеций и дупликаций экзонов 1-10, 21-30, 41-50, 61-70 в гене DMD не обнаружено.
Положительный результат
Обнаружена делеция/дупликация в гене DMD. Диагноз «миодистрофия Дюшенна/Беккера» подтвержден.
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды, индивидуальных генетических факторов. Для оценки характера наследования у детей и родственников, характера развития заболевания в последующем, назначения лечения рекомендуется получить консультацию специалиста.
Скачать пример результатаВажные замечания
- Для получения заключения по результату обследования необходимо проконсультироваться у клинического генетика.
Кто назначает исследование?
Невролог, психиатр, врач-генетик.
Также рекомендуется
[02-006] Общий анализ мочи с микроскопией
[06-022] Креатинкиназа общая
[42-050] Генетическое обследование на болезнь Кеннеди (спинальная и бульбарная мышечная атрофия) в гене AR
Литература
- Aartsma-Rus A, Ginjaar IB, Bushby K The importance of genetic diagnosis for Duchenne muscular dystrophy Journal of Medical Genetics 2016;53:145-151.
- Darras BT, Urion DK, Ghosh PS. Dystrophinopathies. 2000 Sep 5 [Updated 2018 Apr 26]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018.
- Darras BT, Menache-Starobinski CC, Hinton V, Kunkel LM. Chapter 30 — Dystrophinopathies. In: Darras BT, Jones HR, Ryan MM, De Vivo Childhood, and Adolescence (Second Edition) DCBT-ND of I, editors., San Diego: Academic Press; 2015, p. 551–92.
Почему не все врачи знают эту болезнь? Невролог – о миодистрофии Дюшенна
О таком редком и тяжелом генетическом заболевании, как прогрессирующая мышечная дистрофия Дюшенна, практически не говорят в России. Не только родители, но и многие врачи сталкиваются с ним впервые и плохо понимают, что делать с больными детьми. Между тем в настоящее время проводятся десятки международных исследований, направленных на поиски лечения.
О том, как и почему возникает это заболевание, с какими трудностями можно столкнуться при диагностике и существует ли лекарство от страшного недуга, в интервью m24.ru рассказал и.о. руководителя Детского нервно-мышечного центра НИКИ педиатрии кандидат медицинских наук Дмитрий Влодавец.
Фото: m24.ru/Никита Симонов
– Расскажите о заболевании, как оно проявляется и на что родителям стоит обратить внимание, чтобы вовремя его обнаружить?
– По последним данным, с миопатией Дюшенна рождается один из 5000 мальчиков, а не один из 3000, как было принято считать раньше. Если переложить эту статистику на такой крупный город, как Москва, где за год рождаются около 100 тысяч детей, то каждый год должно рождаться 10 детей с миопатией Дюшенна. В среднем болезнь начинает проявляться в возрасте пяти лет. Дети испытывают значительные сложности при ходьбе, быстро устают, им сложно подниматься по лестнице, а при вставании с пола они применяют миопатические приемы Говерса (вставание «лесенкой»). Еще один важный симптом – большие голени. Члены семьи поначалу радуются, думая, что ребенок растет спортсменом, однако вскоре оказывается, что это не так.
– То есть ребенок полностью здоров и вдруг резко в пять лет начинает проявляться заболевание?
– Не совсем. Минимальные проявления обычно встречаются раньше, но могут просто остаться без внимания. Если же родителей подробно расспросить, как ребенок вел себя на детской площадке или во время повседневной двигательной активности, то выясняется, что он, например, так и не научился приседать, или медленно бегал, или не мог подпрыгнуть… Только у 10 процентов пациентов встречается инфантильный тип заболевания, при котором явные клинические проявления возникают с самого рождения. В этом случае ребенок уже на первом году жизни слабый, вялый, позже других начинает ходить, позже приобретает моторные навыки.
– И как в дальнейшем развивается заболевание?
– К сожалению, болезнь довольно быстро прогрессирует. У мальчиков со временем формируется гиперлордоз в поясничном отделе позвоночника (выгибание), возникает остеопороз (снижение плотности костей), контрактуры суставов (ограничение подвижности). В среднем уже в 8–12 лет ребята теряют способность к самостоятельному передвижению. Хотя все зависит от индивидуальных особенностей. Есть мальчики, которые уже в шесть лет садятся в инвалидное кресло, а есть такие, которые ходят и в 15–16 лет.
Когда пациенты теряют возможность самостоятельно передвигаться, у них формируются новые контрактуры, в том числе коленные, тазобедренные, локтевые, межфаланговые. Еще одной проблемой становится искривление позвоночника. Ведь мальчики все равно учатся в школе, что-то пишут, читают, и, если осанка нарушена, у них может сформироваться S-образный сколиоз, который иногда требует хирургического вмешательства.
У 70 процентов пациентов к 15 годам формируется кардиомиопатия, а затем возникает прогрессирующая сердечная и дыхательная недостаточность, отчего они и погибают в возрасте 15–25 лет. Здесь также стоит отметить, что все индивидуально и некоторые пациенты доживают и до 30–40 лет.
Фото: m24.ru/Никита Симонов
– Но почему у детей возникает эта болезнь? Что запускает генетический механизм, приводящий к столь тяжелым последствиям?
– Миодистрофия Дюшенна – это генетическое заболевание, связанное с нарушением синтеза белка дистрофина, который необходим для правильной работы наших мышц. Заболевание наследуется по X-сцепленному типу наследования, так как ген, отвечающий за выработку белка дистрофина, находится на Х-хромосоме. По молекулярным меркам это ген-великан, он состоит из 79 кусочков – экзонов. При наличии мутации в этом гене белок дистрофин в клетках мышц не синтезируется, мышечная ткань постепенно гибнет и замещается жировой и соединительной. В 60 процентах случаев мутация представляет собой делецию (потерю) или дупликацию (удвоение) одного или нескольких экзонов. В остальных случаях мы имеем дело с точечными мутациями.
– А почему миодистрофией Дюшенна болеют только мальчики?
– Дело в том, что в кариотипе мужчины присутствует только одна X-хромосома, которую он получает от матери. И если он получил Х-хромосому с поврежденным геном, то дистрофин у него в организме вырабатываться не будет, соответственно, проявится миодистрофия. У женщины всегда есть две X-хромосомы. И если на одной из них находится больной ген, то вторая здорова и производит дистрофин, поэтому заболевание не проявляется.
Но не всегда мальчик получает больной ген от мамы. Примерно в 40 процентах случаев мутация возникает спонтанно в момент зачатия, при этом ни один родитель не является носителем.
– Я читала, что существует не такая тяжелая форма этого заболевания, которая называется формой Беккера. В чем ее отличие на генетическом уровне?
– Дело в том, что экзоны имеют разную форму. Мы можем представить ген дистрофина как пазл из 79 кусочков, вытянутых в один ряд. Если в гене отсутствуют, например, 51, 52 и 53-й экзоны, то 50-й уже не сможет соединиться с 54-м. Синтез белка начинается, доходит до 50-го экзона и останавливается. Это называется нарушением рамки считывания и вызывает как раз миопатию Дюшенна. Но иногда рамка считывания восстанавливается самой природой. Например, в гене произошла потеря 20-го и 21-го экзонов, но форма 19-го экзона такова, что он может соединиться с 22-м. Синтез белка идет до конца, и получается немного укороченный, но вполне функциональный белок дистрофин. Такой белок тоже работает, и заболевание протекает в более сохранной форме, которая называется формой Беккера. Она встречается реже, примерно у одного на 20 000 новорожденных мальчиков. Заболевание протекает легче, мышечная слабость возникает гораздо позднее. Например, одному из моих пациентов с формой Беккера уже 36 лет и он живет нормальной жизнью. У него есть семья, он водит машину, работает на хорошей работе. Но у этих пациентов может быть более выражена кардиомиопатия. Бывает, что к 18 годам приходится делать пересадку сердца.
– Вернемся к диагностике. Если у родителей есть подозрение на то, что у ребенка миодистрофия Дюшенна, куда им идти и какие сдавать анализы?
– Да, с диагностикой в настоящее время все не так просто. В первую очередь родители обращают внимание на трудности при ходьбе и поэтому идут к ортопедам, а те, как правило, про это заболевание не знают. Так, пока ребенок попадет к неврологу, может пройти несколько лет. Да и не каждый невролог знает это заболевание! Два года назад я стал вести базу наших пациентов. Беру у них кровь, собираю клиническую информацию: что мальчики еще могут делать, чего уже не могут, в каком возрасте садятся в инвалидное кресло. К нам приезжают со всей России и даже из сопредельных государств – Белоруссии, Украины, Киргизии, Казахстана, Таджикистана. Там врачи вообще не знают, что с такими пациентами делать, и очень их боятся. Так вот, сейчас в моей базе 356 пациентов с миодистрофией Дюшенна и Беккера. А по некоторым расчетам, только в России должно быть около 4000 пациентов. Где они? Неизвестно. Врач-невролог по месту жительства может сказать, что ваше заболевание не лечится, ребенок скоро умрет. И родители ничего не делают. Хотя у них есть возможность обратиться в региональное отделение Минздрава – там дают бесплатное направление в Москву на обследование.
Сильно мешает диагностике задержка умственного развития, которая в той или иной степени встречается у 30 процентов пациентов. Например, полгода назад я поставил диагноз девятилетнему пациенту, у которого была очень выраженная задержка развития, и его наблюдали как пациента с аутистическим расстройством.
При миодистрофии Дюшенна креатинкиназа (КФК) в крови повышена в сотни раз! А у нас до сих пор не все врачи знают, что такое анализ КФК. Например, из Тульской области приезжает мальчик со значением КФК 25 единиц. Мы переделываем анализ, и оказывается, что у него на самом деле 25 000 единиц! А часто КФК вообще не смотрят. В основном делают анализы АЛТ и АСТ. Это ферменты, которые в сознании врачей плотно связаны с инфекционными заболеваниями печени – гепатитами, гепатозами, циррозом печени. И, когда врач получает повышенный АЛТ и АСТ, он решает, что у ребенка гепатит. Но в данном случае АЛТ и АСТ имеют внепеченочное происхождение – они выбрасываются в кровь при разрушении мышц.
Фото: m24.ru/Никита Симонов
– Какие методы диагностики должны применяться в первую очередь?
– Можно сделать биопсию мышечной ткани и МРТ мышц. Эти методы позволяют увидеть, что мышечная ткань замещается жировой или на месте мышц разрастаются соединительные ткани.
Но так как мы имеем дело с генетическим заболеванием, для диагностики важно сделать правильный генетический анализ. В российских лабораториях он до сих пор делается методом ПЦР, который позволяет оценить наличие только 19 экзонов. Да, это набор наиболее часто встречающихся мутаций, но не более того. Поэтому к нам приходит очень много пациентов якобы без мутаций. У них есть результат исследования, в котором написано, что мутация не обнаружена. А она у них есть, и, пока идут поиски, заболевание прогрессирует.
Существует современный тест MLPA, который позволяет оценить состояние всех 79 экзонов. Раньше мы сотрудничали с американской лабораторией в Юте, но два года назад сами стали делать его на хорошем уровне, который вполне сопоставим с зарубежными лабораториями.
– Если мутация обнаружена, что делать дальше? Можно ли помочь ребенку, существует ли поддерживающее лечение?
– Во-первых, хорошо себя зарекомендовала гормональная терапия – если вовремя назначить глюкокортикостероиды, то можно добиться пролонгации самостоятельного хождения на два-три года. При регулярном применении снимаются отек и воспаление, связанные с гибелью мышечных клеток, стабилизируется мышечная мембрана, что позволяет сохранить некоторое количество клеток. Обычно назначается один из двух препаратов – преднизолон или дефлазакорт. Дефлазакорт вызывает меньше побочных действий, но пока препарат не зарегистрирован на территории РФ.
Не так давно была доказана эффективность назначения ингибиторов АПФ для профилактики дилатационной кардиомиопатии. Это те препараты, которые обычно пьют бабушки для снижения давления. Однако наши коллеги из Института миологии в Париже провели исследование, которое показало, что при раннем назначении ингибиторов АПФ к 15 годам кардиомиопатия сформировалась всего у 20–30 процентов пациентов, страдающих мышечной дистрофией Дюшенна (вместо 70 процентов, как было раньше). С этой же целью назначаются препараты, снижающие частоту сердечных сокращений.
Для профилактики остеопороза показано назначение препаратов, содержащих витамин D3 и кальций.
Обязательно нужно делать специальные растяжки ежедневно утром и вечером, а на ночь надевать тутора на голеностопные суставы. Это очень важно, однако родители не придают этому значения. На одном приеме у меня был папа больного мальчика – военный из Архангельска. Я показал ему, как делать растяжки, все объяснил. Через год они приезжают – отличное состояние суставов, даже лучше, чем было. Проходит еще год – ребенок продолжает ходить! Оказалось, что папа ежедневно, старательно, в точности выполняет все указания. А кто-то говорит: у нас ребенок хныкал и мы перестали делать. И результат соответствующий…
Часто дети перестают ходить не из-за мышечной слабости как таковой, а из-за жутко запущенных контрактур, с которыми не работают родители или врачи – физические терапевты.
Мнение эксперта
Первая и основная программа фонда – «Мы вместе». Это, например, проведение психолого-реабилитационных лагерей. В семьях, где ребенок с редким диагнозом лишен медицинского сопровождения, встречи с коллективом высокопрофессиональных специалистов трудно переоценить. Но если для москвичей мы регулярно проводим встречи в родительском клубе, то с жителями регионов в этом смысле сложнее. Поэтому дважды в год, осенью и весной, семьи со всей России приезжают на неделю в пансионат в Калужской области. Ребята знакомятся, рукодельничают, проходят тренинги, организуют квесты, а в других аудиториях родители общаются со специалистами. В лагерь приезжают детский и взрослый психологи, невролог, кардиолог, пульмонолог – штучные профессионалы, которых в России можно по пальцам пересчитать. Участие в программе бесплатно для всех семей, в которых есть ребенок с миодистрофией Дюшенна, фонд также берет на себя транспортные расходы.В этом году у нас стартовала медицинская программа «Клиника МДД». Очередь в федеральное лечебное учреждение для наших ребят сейчас составляет примерно год, а для прогрессирующего заболевания – это совершенно неприемлемый срок. Теперь у нас появилась возможность за два дня провести полное обследование тех детей, которые находятся в этом списке ожидания, или подопечных ребят старше 18 лет, которым и обратиться некуда.
В Европе с миодистрофией Дюшенна живут в среднем около 30 лет. К сожалению, в России ребята часто уходят в возрасте двенадцать-четырнадцать-шестнадцать лет из-за того, что им оказывается помощь без учета особенностей заболевания. Например, после потери способности к хождению у ребят быстро слабеют диафрагмальные мышцы, снижается кашлевой рефлекс. В результате ребята не могут самостоятельно кашлять, чтобы откашливать мокроту при простуде или гриппе. И, если врач назначит муколитик, увеличивающий мокроту, ребенок не сможет откашлять ее и начнет задыхаться. В лучшем случае ему сделают дырку в горле и он будет дышать через трахеостому. Еще дадут кислород без контроля газов крови, чем только ухудшат ситуацию. В худшем его не спасут.
Чтобы такого не случалось, медицинскому персоналу необходимо знать особенности этого заболевания и особенности оказания помощи. Семье нужно учиться правильно жить с заболеванием. Респираторные упражнения и физическая терапия, дренажный массаж и откашливатель. В этом залог качества и продолжительности жизни ребят с МДД. Благодаря этому за рубежами нашей родины ребята живут в среднем на 10 лет дольше.
Ребятам нужны индивидуальные ортопедические коляски, регулярный прием лекарств. Все это родители вынуждены оплачивать сами, так как пока в России миодистрофия Дюшенна не имеет стандарта оказания медицинской помощи и ребята лишены системной медицинской помощи. Но сначала необходимы самостоятельные клинические рекомендации по ведению пациентов с мышечной дистрофией Дюшенна. Затем стандарт оказания медицинской помощи. На сегодняшний день это одна из самых главных задач.
Елена Шеперд
Соучредитель фонда для детей с миодистрофией Дюшенна «МойМио»
– Но все же, несмотря на глюкокортикостероиды и растяжки, заболевание прогрессирует и его невозможно остановить. Или возможно? Я знаю, что в настоящее время проходят различные эксперименты, например, испытывается метод лечения под названием экзон-скиппинг. Что это такое и действительно ли он лечит мутированный ген?
– Слово skip по-английски означает «прыжок». Идея заключается в том, что «перепрыгивание» определенных экзонов в гене приводит к восстановлению рамки считывания. А это значит, что в клетках начинает вырабатываться укороченный дистрофин и болезнь переходит в более сохранную форму Беккера.
Две зарубежные компании – Prosensa (Нидерланды) и Sarepta (США) – проводили клинические испытания экзон-скиппинга 51-го экзона, под которые подходили пациенты с определенными делециями – приблизительно 13 процентов Дюшеннов. Мы принимали участие в третьей фазе клинических испытаний: из 186 пациентов со всего мира восемь были нашими. Раз в неделю в течение нескольких лет мальчикам делалась подкожная инъекция. Однако после анализа данных оказалось, что в результате исследований не было получено никакой статистически достоверной разницы между теми пациентами, которые получали лечение, и теми, которые получали плацебо. Фирма Prosensa, которая разрабатывала препарат, обанкротилась. Сейчас эти исследования и клинические испытания по экзон-скиппингу продолжает американская компания Sarepta.
Теоретически этот метод подходит только для пациентов с часто встречающимися делециями, что составляет примерно половину больных мальчиков. Разработка одного препарата для экзон-скиппинга стоит миллиарды долларов, и для пациентов с редкими делециями ее, конечно, делать не будут.
Фото: m24.ru/Никита Симонов
– Я слышала также, что уже существует препарат «Трансларна», который очень дорого стоит и в России не продается.
– Да, аталурен, или «Трансларна», подходит только для пациентов с точечными стоп-мутациями. Он способен отыскивать неправильно возникший стоп-сигнал и прочитывать ген сквозь него. Препарат действительно очень дорогой: курс лечения на ребенка весом 25 килограммов в год составляет около 600 тысяч евро.
– И есть люди, которые его покупают?
– Со слов компании «PTC», производящей препарат, его принимают до тысячи человек. В США он пока не одобрен, а в Евросоюзе получил одобрение с условием, что компания проведет еще одно клиническое испытание. В Россию компания пока не обращалась за регистрацией. Даже если они проявят желание и подадут документы, этот процесс может занять несколько лет. В России есть несколько пациентов, которым препарат «Трансларна» был закуплен при помощи благотворительных фондов. Пока же планируется клиническое исследование, куда мы постараемся включить максимально возможное количество наших пациентов.
– Какие еще испытания проводятся сейчас в мире?
– Их очень много. Во французском Институте миологии сейчас ведутся испытания на животных, которых заражают вирусными векторами, несущими микродистрофин. Эту генетическую структуру сажают на аденовирус, из которого предварительно выделяют все паталогические ДНК, и затем он должен заразить каждую клеточку организма, чтобы в ней начался синтез дистрофина. Проблема в том, что аденовирус не способен заключить в себя всю нуклеотидную последовательность гена – настолько она огромная. Поэтому может использоваться только микроген, что позволит лишь перевести форму Дюшенна в более сохранную форму Беккера, а не полностью восстановить синтез дистрофина. Но МРТ и биопсии мышц показывают, что в результате исследований у лабораторных животных некоторые мышечные клетки действительно начинают вырабатывать дистрофин, и это очень хорошо.
Есть и гипотезы, связанные со стволовыми клетками. Делая МРТ мышц, мы видим, что до пяти-шести лет у пациентов с миодистрофией Дюшенна мышцы не изменены. Возможно, это происходит за счет работы стволовых клеток. Когда мышечная клетка гибнет, на ее место приходит стволовая, пытается заместить ее и как-то работать. И так происходит, пока запас стволовых клеток не истощается. Но пока это только гипотеза.
Существует идея использования «уснувшего» гена – утрофина. Это такой эмбриональный дистрофин – он работает, только когда плод находится в эмбриональном состоянии, а затем инактивируется. Если каким-то образом снять блокировку и вновь заставить его функционировать, то он вполне может замещать неполноценный белок дистрофин и восстанавливать нормальную работу мышц.
Фото: m24.ru/Никита Симонов
– А какие исследования проводятся в России?
– К сожалению, у нас наблюдается гигантский провал в этом научном направлении. Если государство не тратит ничего на свою науку, то потом придется тратить огромные деньги за чужую. И в 1990-е, и в 2000-е годы, и до сих пор на изучение нервно-мышечных заболеваний не выделяется никаких бюджетных денег. Наше отделение из 30 коек целиком создано на энтузиазме сотрудников. Но мы клиницисты, мы не можем в стационаре разрабатывать препараты, работать с молекулами, лабораторными животными. Это работа для молекулярных биологов, ветеринаров, фармацевтов, провизоров-технологов и других. И только после того как будет доказана безопасность испытываемой молекулы-препарата, проводятся клинические испытания первой, второй и третьей фазы, в которых уже могут принимать участие пациенты.
– Кто же оплачивает зарубежные исследования?
– Например, во Франции Институт миопатии живет только за счет пожертвований. Ежегодно в декабре родители детей с нервно-мышечными заболеваниями устраивают мощную благотворительную акцию «Телетон», в которой принимают участие звезды шоу-бизнеса, популярные телеведущие, актеры. Каждый желающий может в прямом эфире позвонить и пожертвовать любую сумму на исследования. В результате ежегодно они собирают миллионы евро! Их примеру последовали в США, Великобритании, Италии. У нас в этом смысле больше распространена адресная помощь. Люди собирают деньги на конкретную операцию, покупку кресла для конкретного ребенка, но не на научные исследования. В России априори считается, что всю научную деятельность ведут государственные НИИ и там что-то наверняка делается, хотя это совсем не так.
Единственные российские научные исследования по Дюшенну, которые сейчас ведутся, – это малоизвестный частный проект, у которого только один спонсор – отец больного мальчика.
Мнение эксперта
Наша компания возникла три года назад, когда у сына моего знакомого диагностировали миодистрофию Дюшенна. Выяснилось, что заболевание никак не лечат, есть лишь методы поддерживающей терапии, но прогноз все равно неблагополучный.Экзон-скиппинг 51-го экзона, который в тот момент разрабатывали Prosensa и Sarepta, не подходил нашему пациенту. Мы решили сделать это по-другому, повторяли описанные эксперименты раз за разом, но смогли убедиться только в том, что этот подход не работает, соответственно, надо искать новый.
Полтора года назад благодаря новым технологиям стало возможно доставлять в клетки генные конструкции с помощью аденоассоциированных вирусов. Сейчас мы разрабатываем препарат, который будет доставлять микроген в клетки. Пока проходят эксперименты на мышах, и, как только мы поймем, что технология работает, начнем синтезировать вирусы в больших количествах и приступим к доклиническим исследованиям.
Конечно, мы не единственная организация, которая развивает вирусный подход. Но обычно делают упор на микродистрофин, а мы больше работаем с микроутрофином. По нашим ожиданиям, этот подход подойдет всем пациентам, независимо от вида мутации.
Денис Решетов
Биоинженер и биоинформатик, директор по науке компании «Марлин Биотех»
– Вам психологически легко работать с больными детьми, зная о том, что их ждет?
– Конечно, детей очень жалко, но кто-то же должен им помогать. Когда я был молодым специалистом, то одно время думал, что схожу с ума. Было такое впечатление, что все дети вокруг больные. Тогда я устроился в поликлинику на четверть ставки, ведь там на приеме были одни здоровые дети! Легко и приятно смотреть на здоровых детей! И постепенно в голове все устаканилось.
На самом деле гораздо труднее общаться с родителями. У наших родителей первая реакция – неприятие. Они не верят диагнозу, считают его ошибочным, некоторые едут перепроверяться в Израиль или США. Думаю, это срабатывает механизм психологической защиты. А вот с теми родителями, которые принимают заболевание, работать уже значительно легче.
– Если бы у вас была возможность перебраться за рубеж, вы бы ею воспользовались?
– Такая возможность была, но все-таки только здесь я ощущаю себя на своем месте. Да, существуют различные ограничения, но я делаю все, что от меня зависит. Принимаю пациентов, читаю лекции студентам на кафедре неврологии, нейрохирургии и медицинской генетики педиатрического факультета РНИМУ имени Н.И. Пирогова. И мечтаю, что когда-нибудь и в России появится нервно-мышечный центр.
Полезная информация
Записаться на прием, проконсультироваться о заболевании:Дмитрий Влодавец,
и.о. руководителя Детского нервно-мышечного центра НИКИ педиатрии имени Ю. Вельтищева,
к.м.н., доцент кафедры неврологии, нейрохирургии и медицинской генетики РНИМУ имени Н.И. Пирогова.
Тел.: 8 (905) 744-61-03
E-mail: [email protected]
Узнать о ходе исследований, оказать финансовую поддержку:
Денис Решетов,
биоинженер и биоинформатик,
директор по науке компании «Марлин Биотех».
Тел.: 8 (917) 523-26-84
E-mail: [email protected]
Сайт: gendeti.com
Для получения поддержки и участия в работе фонда, а также для оказания финансовой поддержки:
Благотворительный фонд помощи детям с миодистрофией Дюшенна и их семьям «МойМио»
Тел.: 8 (495) 055-61-95
E-mail: [email protected]
Сайт: mymiofond.ru
Алена Водопьянова
БЕККЕРА СИНДРОМ — это… Что такое БЕККЕРА СИНДРОМ?
- БЕККЕРА СИНДРОМ
- (Беккера – Рейтера синдром, описан американским дерматологом S. W. Becker, 1894–1964, с соавт.) – наследственный симптомокомплекс: множественные слабопигментированные пятна с атрофией эпидермиса на коже подбородка, шеи и плеч, появляющиеся в раннем детстве; пятна становятся более заметными во время нервного возбуждения. Тип наследования не установлен. Возможна косметическая коррекция.
S. W. Becker, M. J. Reuter. Familial pigmentary anomaly. Archives of Dermatology and Syphilology, Chicago, 1939; 40: 987–998.
Энциклопедический словарь по психологии и педагогике. 2013.
- БЕККЕРА СИМПТОМ
- Белара-Дежана-Пе синдром
Смотреть что такое «БЕККЕРА СИНДРОМ» в других словарях:
Синдром Шарко–Вейса–Беккера — См. Синдром каротидного синуса … Энциклопедический словарь по психологии и педагогике
Беккера-Мьюэра синдром — (S. W. Becker, род. в 1894 г., амер. дерматолог; К. В. Muir) см. Псевдоатрофодерма шеи … Большой медицинский словарь
Беккера-Рейтера синдром — (S. W. Becker, род. в 1894 г., амер. дерматолог; P. Reuter, род. в 1907 г., нем. дерматолог) наследственный симптомокомплекс: наличие на коже подбородка, шеи и плеч пигментных пятен с атрофией эпидермиса … Большой медицинский словарь
Синдром каротидного синуса Шарко–Вейса–Беккера — При раздражении каротидного синуса возникают головокружение, шум в голове, расстройство зрения на той же стороне, чувство страха, одышка, боли в области сердца, нарушение сердечного ритма, иногда потеря сознания и судорожные подергивания на… … Энциклопедический словарь по психологии и педагогике
МКБ-10: Класс VI — Список классов Международной классификации болезней 10 го пересмотра Класс I. Некоторые инфекционные и паразитарные болезни Класс II. Новообразования Класс III. Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный… … Википедия
МКБ-10: Класс G — Список классов Международной классификации болезней 10 го пересмотра Класс I. Некоторые инфекционные и паразитарные болезни Класс II. Новообразования Класс III. Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный… … Википедия
МКБ-10: Код G — Список классов Международной классификации болезней 10 го пересмотра Класс I. Некоторые инфекционные и паразитарные болезни Класс II. Новообразования Класс III. Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный… … Википедия
ДИСТРОФИЯ МЫШЕЧНАЯ ДЮШЕННА — мед. Мышечная дистрофия Дюшенна наследственная прогрессирующая мышечная дистрофия, характеризующаяся началом в раннем возрасте, симметричной атрофией мышц в сочетании с сердечно сосудистыми, костно суставными и психическими нарушениями,… … Справочник по болезням
Дилатационная кардиомиопатия — МКБ 10 I42.042.0 МКБ 9 425.4425.4 OMIM … Википедия
Дистрофи́и мы́шечные прогресси́рующие — (синоним миодистрофии) группа наследственно обусловленных нервно мышечных заболеваний, характеризующихся прогрессирующей мышечной слабостью, атрофией мышц, двигательными нарушениями. Различные формы Д. м. п. отличаются разным типом наследования,… … Медицинская энциклопедия
Мышечная дистрофия Дюшенна / Беккера — ДНК-диагностика
Миодистрофия Дюшенна (МДД) — наследственное заболевание, которое начинается в возрасте 2-5 лет и характеризуется прогрессирующеймышечной слабостью, атрофией и псевдогипертрофией проксимальных мышц, нередко сопровождается кардиомиопатиями и нарушением интеллекта. На ранних этапах заболевания наблюдается повышенная утомляемость при ходьбе, изменение походки («утиная походка»). При этом происходит постепенная деградация мышечных тканей. 95% больных перестают ходить в возрасте 8-12 лет. В возрасте 18-20 лет больные, как правило, умирают, часто от дыхательной недостаточности. Выделяют аллельную МДД форму – мышечную дистрофию Беккера (МДБ, OMIM 310200), которая характеризуется сходными клиническими проявлениями, более поздним началом (примерно в 10-16 лет) и более мягким течением. Такие больные часто сохраняют способность ходить до 20 лет, а некоторые – до 50-60 лет, хотя в патологический процесс вовлечены те же мышцы, что и при МДД. Продолжительность жизни таких больных сокращена незначительно.
Биохимическим маркером заболевания является повышенный (в 100-200) раз уровень креатинфосфокиназы (КФК) в крови. У носительниц поврежденного гена уровень КФК в среднем также несколько повышен.
Тип наследования мышечной дистрофии Дюшенна – Х-сцепленный рецессивный, т.е. им страдают почти исключительно мальчики, женщины же с поврежденным геном в одной из Х-хромосом являются носительницами МДД. Но в редких случаях миодистрофией Дюшенна могут болеть и девочки. Причинами этого могут быть преимущественная инактивация Х-хромосомы с нормальным аллелем у гетерозиготных носительниц мутантного гена DMD, Х-аутосомная транслокация, затрагивающая этот ген, гемизиготность по мутантному аллелю и наличие фенокопий (заболеваний, связанных с нарушением других белков, входящих в дистрофин-гликопротеиновый комплекс). Приблизительно в 2/3 случаев сын получает хромосому с повреждением от матери-носительницы, в остальных случаях заболевание возникает в результате мутации de novo в половых клетках матери или отца, либо в предшественниках этих клеток. Мышечная дистрофия Дюшена (МДД) встречается приблизительно у одного из 2500-4000 новорожденных мальчиков.
Ген DMD, ответственный за прогрессирующую мышечную дистрофию Дюшена/Беккера (МДД/МДБ), находится в локусе Хр21.2, имеет размер 2,6 млн п.н. и состоит из 79 экзонов. В 60% случаев мутации, приводящие к МДД/МДБ, представляют собой протяженные делеции (от одного до до нескольких десятков экзонов), в 30% случаев – точковые мутации и в 10% случаев – дупликации. Из-за наличия так называемых «горячих участков» делеций амплификация 27 экзонов и промоторной области гена DMD позволяет выявлять примерно 98% всех крупных делеций. Поиск точковых мутаций затруднен из-за большого размера гена и отсутствия мажорных мутаций.
В Центре Молекулярной Генетики проводится измерение уровня КФК в крови, а также прямая диагностика МДД/МДБ, представляющая собой поиск крупных делеций/lдупликаций во всех экзонах гена DMD и поиск «точковых» мутаций гена DMD методом NGS (next generation sequensing). Исследование методом NGS позволяет так же выявлять делеции всех экзонов гена DMD у больных мальчиков. Анализ всех экзонов гена позволяет определить точные экзонные границы делеции в сучае ее выявления, и таким образом, установить приводит ли данная делеция к сдвигу рамки считывания белка, что в свою очередь важно для прогноза формы заболевания — миодистрофия Дюшенна или Беккера. Таким образом, сочетание различных методов исследования позволяет выявлять практически все мутации гена DMD.
Наличие любого типа мутаций (делеции/дупликации в одном или нескольких экзонах, «точковые» мутации) является молекулярно-генетическим подтверждением клинического диагноза миодистрофии Дюшена/Беккера и позволяет проводить дородовую диагностику в данной семье.
Внимание! Для измерения уровня КФК кровь должна быть свежей (не замороженной)!
В случае дородовой диагностики необходим биоматериал плода, в качестве которого можно использовать ворсины хориона (с 8-й до 12-й недели беременности), амниотическую жидкость (с 16-й до 24-й недели беременности) или пуповинную кровь (с 22-й недели беременности).
Нами разработаны наборы для ДНК-диагностики прогрессирующей мышечной дистрофии Дюшенна / Беккера. Наборы предназначены для использования в диагностических лабораториях молекулярно-генетического профиля.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий — около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Публикации по теме раздела
Мышечная дистрофия Дюшенна-БеккераМиодистрофия Беккера
Миодистрофия Беккера – наследственное заболевание мышечной ткани, для которого характерно развитие одного или более дефектных генов, которые могут привести к мышечной слабости различной степени. Данная патология вызывает ослабление мышц, близко расположенных к телу. Врач ставит такой диагноз после проведения диагностическое обследования. Недуг встречается крайне редко у детей.
Детский врач клиники Tel Aviv Medical Clinic более 10 лет специализируется на лечение различных патологий у детей, в том числе миодистрофии Беккера. С помощью современного оборудования удается выявить опасное заболевание на ранней стадии. Мы используем инновационные методики терапии. Наш медперсонал ежегодно проходит специальное обучение и знает, как наладить дружеский контакт с детьми. Наши сотрудники установили тарифы на уровне государственных. Это делает предоставление наших услуг более доступным для различных слоев населения.
Как проявляется патология
Родители малыша могут заметить проявление данного заболевания в подростковом возрасте. Тревожными симптомами могут быть:
- Проблемы с сердцем;
- Нарушение дыхания;
- Задержка развития;
- Слабость плечевых мышц;
- Плохая работа суставов;
- Увеличение икроножных мышц;
- Частая пневмония;
- Невозможность двигаться.
Зачастую заболевание возникает у мужского пола. Для постановки верного диагноза врач проводит комплексное обследование организма.
Лечение патологии в клинике Tel Aviv Medical Clinic
У нас работает настоящая команда профессионалов, которая проводит диагностику и лечение. Мы используем комплексный подход, который позволяет избавить ребенка от причины возникновения заболевания и подобрать индивидуальную схему терапии. Наш медперсонал обеспечивает маленьким пациентам круглосуточную заботу и контроль. Мы предоставляем комфортабельные палаты для совместного пребывания мамы и малыша.
В нашей клинике есть центр поддержки родителей и детей, в котором можно получить квалифицированную помощь психологов и логопедов. Наш медперсонал помогает связаться с международными благотворительными фондами для сбора средств на лечение. Врачам клиники удается помочь пациентам, которых признали безнадежными в других больницах стран СНГ.
Мышечная дистрофия Беккера (МПК) — Заболевания
Мышечная дистрофия Беккера (BMD)
Что такое мышечная дистрофия Беккера?
Мышечная дистрофия Беккера (BMD ) — один из девяти типов мышечных дистрофий, группы генетических дегенеративных заболеваний, в первую очередь затрагивающих произвольные мышцы. BMD относится к группе дистрофинопатий, включая мышечную дистрофию Дюшенна (DMD) и промежуточную форму между DMD и BMD.Заболевание названо в честь немецкого врача Питера Эмиля Беккера, который впервые описал этот вариант МДД в 1950-х годах. МПК похожа на МДД, но позволяет произвольным мышцам функционировать лучше, чем при МДД. BMD имеет более позднее начало и более легкие симптомы по сравнению с DMD. Однако сердечная мышца может быть поражена так же, как при МДД.
Каковы симптомы МПК?
Начало BMDшироко варьирует от 5 до 60 лет, 1 и течение более медленное и менее предсказуемое, чем у DMD.Общая слабость сначала поражает мышцы бедер, тазовой области, бедер и плеч. Телята часто бывают увеличены. Может быть значительное поражение сердца. Для получения дополнительной информации см. Признаки и симптомы.
Что вызывает МПК?
До 1980-х годов было мало что известно о причинах мышечной дистрофии. В 1986 году исследователи, поддерживаемые MDA, определили ген, который в случае дефекта — проблема, известная как мутация — вызывает DMD и BMD. В 1987 году белок, связанный с этим геном, был идентифицирован и назван дистрофин .
Гены содержат коды или рецепты белков, которые являются важными биологическими компонентами во всех формах жизни. МПК возникает, когда белок дистрофин, который производится из определенного гена на Х-хромосоме, функционирует только частично.
Белок дистрофин предохраняет мембрану мышечной клетки от разрыва или разрыва, когда наши мышцы сокращаются и расслабляются. Поскольку он соединяет центр мышечной клетки с периферией, белок дистрофин очень длинный. Один конец предназначен для соединения с внутренней частью мышцы, а другой конец — для соединения с различными белками на клеточной мембране.Длинная средняя часть, называемая стержневым доменом, занята серией повторяющихся единиц, называемых спектриновыми повторами.
Повторяющиеся спектриновые единицы в середине белка играют важную роль в связывании двух концов, но белок все еще может функционировать (хотя и не идеально) с меньшим количеством из них, чем обычно. Мутации, вызывающие МПК, уменьшают количество этих повторов, что приводит к мышечной слабости.
В дополнение к своей роли передачи силы, дистрофин обеспечивает основу для удержания множества молекул на месте рядом с клеточной мембраной.Потеря дистрофина вытесняет эти молекулы с последующим нарушением их функций.
В то время как мутации DMD фактически не вызывают образования функционального дистрофина, люди с BMD вырабатывают частично функциональный дистрофин. Они производят сокращенную форму белка, которая защищает мышцы людей с МПК от дегенерации так же полностью или так же быстро, как у людей с МДД.
BMD в первую очередь поражает мальчиков и мужчин, которые наследуют болезнь от своих матерей.Женщины могут быть носителями, но обычно не проявляют никаких симптомов. Подробнее о том, как мутации генов вызывают МПК, см. Причины / Наследование.
Какова продолжительность жизни при МПК?
Большинство людей с МПК доживают до среднего и позднего возраста. Для получения дополнительной информации о жизни с BMD см. Медицинский менеджмент. Если сердечные аспекты заболевания минимальны или если они адекватно контролируются с помощью медицинского вмешательства, можно ожидать нормальной или почти нормальной продолжительности жизни. Пациенты с МПК обычно живут не менее 30 лет.У них средний возраст смерти около 40 лет. Основная причина смерти пациентов с МПК — сердечная недостаточность в результате дилатационной кардиомиопатии.
Каков статус исследований МПК?
Исследователи активно реализуют несколько стратегий в области ПРО. Среди основных стратегий — замена генов, модификация генов, использование стволовых клеток, ингибирование белка под названием миостатин, расширение распределения и повышение уровня белка под названием утрофин, и увеличение притока крови к мышцам.
MDA поддерживает исследования в нескольких из этих областей. Некоторые из этих стратегий проходят испытания на людях. Подробности см. В разделе «Исследования».
Загрузите наш информационный бюллетень о мышечной дистрофии Беккера (BMD)
Ссылки
- Брэдли, У. Г., Джонс, М. З., Муссини, Дж. -М и Фосетт, П. Р. У. Мышечная дистрофия по типу Беккера. Мышечный нерв (1978). DOI: 10.1002 / mus.880010204
Мышечная дистрофия Беккера | Johns Hopkins Medicine
Каковы признаки и симптомы мышечной дистрофии Беккера?
Признаки и симптомы мышечной дистрофии Беккера проявляются у пациентов в подростковом или молодом возрасте.Как и в случае более серьезной мышечной дистрофии Дюшенна, мышечное ослабление и истощение обычно начинается в области бедер и таза, а затем переходит на бедра и плечи.
По мере ослабления мышц пациенты могут замечать изменения, когда они занимаются физическими упражнениями и спортом. Эта слабость может вызвать изменение походки. Люди, страдающие мышечной дистрофией Беккера, могут начать ковылять, ходить на носках или толкать живот вперед при ходьбе, чтобы поддерживать равновесие и компенсировать недостаток силы в бедрах и ногах.
Каковы факторы риска мышечной дистрофии Беккера?
Мышечная дистрофия Беккера — это генетическое заболевание, вызванное геном на Х-хромосоме, который матери, несущие этот ген, могут передать своим сыновьям.
Как диагностируется мышечная дистрофия Беккера?
Диагностика мышечной дистрофии Беккера сложна, поскольку она имеет много общих симптомов с другими заболеваниями, включая мышечную дистрофию Дюшена, пояснично-конечностную мышечную дистрофию и спинальную мышечную атрофию.
Задача состоит в том, чтобы определить, происходит ли слабость в самих мышцах или в двигательных нейронах (ответвляющихся от спинного мозга), которые контролируют эти мышцы.
Тщательное медицинское обследование, а также наличие признаков и симптомов в анамнезе — это первый шаг, чтобы врач мог отметить характер прогрессирования. Диагностические тесты для мышечной дистрофии Беккера включают:
Анализы крови: Генетические анализы крови могут выявить мутацию гена, ответственного за мышечную дистрофию Беккера. Они также могут измерить присутствие креатинкиназы, фермента, который образуется при разрушении мышечной ткани. Это вещество повышается при мышечной дистрофии и воспалительных заболеваниях.
Биопсия мышц: для тех детей, у которых есть клинические признаки мышечной дизитрофии Дюшенна, но у которых нет ни одной из распространенных мутаций, берут небольшой образец мышечной ткани и исследуют под микроскопом для подтверждения диагноза.
Электромиограмма: этот тест проверяет, является ли мышечная слабость результатом разрушения мышечной ткани, а не повреждения нервов.
Электрокардиограмма (ЭКГ или ЭКГ): тест, который регистрирует электрическую активность сердца, ЭКГ показывает аномальные ритмы (аритмии или аритмии) и выявляет повреждение сердечной мышцы.
Сердце состоит в основном из мышц, поэтому оно поражено мышечной дистрофией. Мышечная дистрофия Беккера может вызвать кардиомиопатию, ослабление сердечных мышц, что, если его не лечить, может привести к сердечной недостаточности и необходимости трансплантации.
Лечение мышечной дистрофии Беккера
В настоящее время не существует лекарства от мышечной дистрофии Беккера. Врач может назначить стероидные препараты, чтобы помочь людям сохранять способность ходить как можно дольше.
Клиническое течение мышечной дистрофии Беккера варьирует. Некоторым людям может потребоваться инвалидная коляска к 30 годам; другие могут продолжать ходить с тростью или без нее в течение многих лет.
Многопрофильная группа специалистов с опытом лечения мышечной дистрофии Беккера может помочь устранить симптомы:
Специалисты по физической и профессиональной реабилитации могут разрабатывать программы упражнений и обучать упражнениям на растяжку, чтобы минимизировать контрактуры, которые представляют собой затвердевшие или деформированные суставы, вызванные сокращением мышц и сухожилий.
Хирурги-ортопеды, специализирующиеся в области мышечной дистрофии, могут лечить контрактуры и сколиоз.
Кардиологи отслеживают сердечную функцию пациента с помощью ЭКГ и эхокардиограммы.
Мышечная дистрофия Беккера: основы практики, патофизиология, эпидемиология
Беккер П.Е., Кинер Ф. [Новая х-хромосомная мышечная дистрофия.]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr .1955. 193 (4): 427-48. [Медлайн].
Becker PE. Два семейства доброкачественной рецессивной мышечной дистрофии, связанной с полом. Рев. Банка Биол . 1962 сентябрь-декабрь. 21: 551-66. [Медлайн].
Andrews JG, Wahl RA. Мышечная дистрофия Дюшенна и Беккера у подростков: современные перспективы. Средство для здоровья подростков Ther . 2018. 9: 53-63. [Медлайн]. [Полный текст].
Thada PK, Bhandari J, Umapathi KK. Мышечная дистрофия Беккера. StatPearls . 2021 Январь [Medline]. [Полный текст].
Angelini C, Fanin M, Pegoraro E, et al. Клинико-молекулярная корреляция у 104 пациентов с легкой Х-сцепленной мышечной дистрофией: характеристика субклинических фенотипов. Нервно-мышечное расстройство . 1994 июл.4 (4): 349-58. [Медлайн].
Гурвич О.Л., Туохи Т.М., Ховард М.Т. и др. Мутации псевдоэксона МДД: эффективность сплайсинга, фенотип и потенциальная терапия. Энн Нейрол .2008, январь 63 (1): 81-9. [Медлайн].
Эштон Э.Дж., Яу С.К., Динз З.С. и др. Одновременное сканирование мутаций на предмет грубых делеций, дупликаций и точечных мутаций в гене DMD. евро J Hum Genet . 2008 16 января (1): 53-61. [Медлайн].
Arahata K, Beggs AH, Honda H и др. Сохранение С-конца молекулы дистрофина в скелетных мышцах от мышечной дистрофии Беккера. Дж. Neurol Sci . 1991 Февраль 101 (2): 148-56.[Медлайн].
Кениг М., Беггс А.Х., Мойер М. и др. Молекулярная основа мышечной дистрофии Дюшенна и Беккера: корреляция степени тяжести с типом делеции. Ам Джам Генет . 1989 Октябрь 45 (4): 498-506. [Медлайн]. [Полный текст].
Schwartz M, Hertz JM, Sveen ML, et al. LGMD2I с характерным фенотипом мышечной дистрофии Дюшенна или Беккера. Неврология . 2005 10 мая. 64 (9): 1635-7. [Медлайн].
Cirak S, Feng L, Anthony K, Arechavala-Gomeza V, Torelli S, Sewry C и др. Восстановление дистрофин-ассоциированного гликопротеинового комплекса после терапии пропуска экзона при мышечной дистрофии Дюшенна. Мол тер . 2011 15 ноября. [Medline].
Mavrogeni S, Markousis-Mavrogenis G, Papavasiliou A, Kolovou G. Поражение сердца при мышечной дистрофии Дюшенна и Беккера. Мир J Кардиол . 2015 26 июля. 7 (7): 410-4.[Медлайн].
Николас А., Рагуэн-Николь С., Бен Яу Р. и др. Выраженность мышечной дистрофии Беккера связана со структурой дистрофина. Хум Мол Генет . 2015 г., 1. 24 (5): 1267-79. [Медлайн].
Наблюдение за здоровьем сердечно-сосудистой системы для лиц, страдающих мышечной дистрофией Дюшенна или Беккера. Педиатрия . 2005 декабрь 116 (6): 1569-73. [Медлайн]. [Полный текст].
Эмери А.Е., Скиннер Р.Клинические исследования доброкачественной (тип Беккера) Х-сцепленной мышечной дистрофии. Клин Генет . 1976 Октябрь 10 (4): 189-201. [Медлайн].
Holloway SM, Wilcox DE, Wilcox A, et al. Ожидаемая продолжительность жизни и смерть от кардиомиопатии среди носителей мышечной дистрофии Дюшенна и Беккера в Шотландии. Сердце . 11 октября 2007 г. [Medline].
Young HK, Barton BA, Waisbren S, et al. Когнитивный и психологический профиль мужчин с мышечной дистрофией Беккера. J Детский Neurol . 2007 г. 3 декабря [Medline].
De Wel B, Willaert S, Nadaj-Pakleza A, et al. Снижение дыхания у взрослых пациентов с мышечной дистрофией Беккера: продольное исследование. Нервно-мышечное расстройство . 2020 26 декабря. [Medline].
Zhang H, Zhu Y, Sun Y, et al. Уровень креатинина в сыворотке: дополнительный показатель, позволяющий отличить мышечную дистрофию Дюшенна от мышечной дистрофии Беккера. Маркер Дис . 2015 г.2015: 141856. [Медлайн].
Menezes MP, North KN. Унаследованные нервно-мышечные расстройства: путь к диагностике. J Детский педиатр . 2011 г. 3 ноября [Medline].
Лим BC, Ли С., Шин Дж.Й., Ким Джи, Хван Х, Ким К.Дж. и др. Генетическая диагностика мышечной дистрофии Дюшенна и Беккера с использованием технологии секвенирования нового поколения: комплексный поиск мутаций на единой платформе. Дж. Мед Генет . 2011 ноябрь 48 (11): 731-6.[Медлайн].
Марти Б., Жиль Р., Туссент М. и др. Комплексная оценка структурных и функциональных нарушений миокарда при мышечной дистрофии Беккера с использованием количественной магнитно-резонансной томографии сердца. Eur Heart J Cardiovasc Imaging . 24 декабря 2018 г. [Medline].
Руткове С.Б., Даррас БТ. Электроимпедансная миография для оценки детей с мышечной дистрофией: предварительное исследование. Журнал Физ. Конф., Сер. .2013. 434 (1): [Medline]. [Полный текст].
Grootenhuis MA, de Boone J, van der Kooi AJ. Жизнь с мышечной дистрофией: последствия для качества жизни, связанные со здоровьем, для детей и взрослых. Результаты здорового образа жизни . 2007. 5:31. [Медлайн]. [Полный текст].
Ямада Ю., Каваками М., Вада А., Оцука Т., Мураока К., Лю М. Сравнение дисфункции глотания при мышечной дистрофии Беккера и мышечной дистрофии Дюшенна. Дезабил Рехабил .2017 13 марта. 1-5. [Медлайн].
Hayes J, Veyckemans F, Bissonnette B. Мышечная дистрофия Дюшенна: возвращение к старой проблеме анестезии. Педиатр Анаэст . 2008 18 февраля (2): 100-6. [Медлайн].
Segura LG, Lorenz JD, Weingarten TN, Scavonetto F, Bojanic K, Selcen D, et al. Анестезия и мышечная дистрофия Дюшенна или Беккера: обзор 117 воздействий анестетика. Педиатр Анаэст . 2013 Сентябрь 23 (9): 855-64. [Медлайн].
Дженкинс Х.М., Стоки А., Криеллаарс Д., Пастеркамп Х. Накладывание дыхания у детей с нервно-мышечными расстройствами. Педиатр Пульмонол . 2013 16 августа [Medline].
Pouwels S, de Boer A, Leufkens HG, Weber WE, Cooper C, van Onzenoort HA, et al. Риск перелома у пациентов с мышечными дистрофиями. Остеопорос Инт . 2013 15 августа [Medline].
Дуан Д. Myodys, полноразмерный плазмидный вектор дистрофина для генной терапии мышечной дистрофии Дюшенна и Беккера.Номер Curr Opin Mol Ther . 2008 10 февраля (1): 86-94. [Медлайн].
Bowles DE, McPhee SW, Li C, Gray SJ, Samulski JJ, Camp AS, et al. Фаза 1 Генная терапия мышечной дистрофии Дюшенна с использованием оптимизированного для трансляции вектора AAV. Мол тер . 2011 8 ноября [Medline].
Мышечная дистрофия Дюшенна и Беккера (DMD / BMD)
Мышечная дистрофия Дюшенна — одно из наиболее распространенных наследственных заболеваний во всем мире.
Это заболевание поражает почти исключительно мальчиков. Родители могут сначала увидеть, что их ребенок от трех до пяти лет часто падает, медленно бегает, ходит на цыпочках или переваливается. Икры у ребенка часто бывают необычно большими. Первоначально слабость наиболее выражена в мышцах бедер и верхней части ног, но со временем она будет распространяться на большинство произвольных мышц, включая те, которые отвечают за дыхание. Сердце тоже со временем слабеет. Из-за слабости сердца и дыхательных мышц это заболевание становится смертельным и требует тщательного медицинского лечения.Мышечная дистрофия Беккера похожа на мышечную дистрофию Дюшена в том, что она может вызывать слабость скелетных мышц, мышц дыхания и сердца, но симптомы обычно менее серьезны.
Диагноз:
Мышечная дистрофия Дюшенна и Беккера вызывается мутациями белка, называемого дистрофином. При мышечной дистрофии Дюшенна функционирующий дистрофин полностью отсутствует в мышцах, тогда как при мышечной дистрофии Беккера присутствует некоторое количество дистрофина, хотя его недостаточно для полностью нормальной функции мышц.
Диагноз ставится на основании истории болезни, включая семейный анамнез, клинических признаков, обнаруженных во время обследования, и подтверждающих лабораторных данных. Мутации в гене дистрофина можно обнаружить с помощью генетического анализа крови. В большинстве случаев мышечной дистрофии Дюшенна и Беккера мутация наследуется от матери ребенка, которая несет ген болезни, но не имеет симптомов; в других случаях мутация происходит спонтанно. Наш генетический консультант внимательно изучит историю болезни в каждой семье, обсудит принципы наследования и поможет взвесить риски и преимущества генетического тестирования различных членов семьи, включая пациента и мать.
Обращение:
Лечение МДД / МПД требует активного участия медицинских работников, родителей и школ для обеспечения благополучия каждого ребенка. Большинству детей с МДД назначают кортикостероиды, которые, как показали клинические испытания, снижают скорость снижения силы. Невролог из нашей команды проведет это лечение и поможет свести к минимуму побочные эффекты препарата. Невролог также направляет и координирует оказание помощи среди множества служб. Физическая и профессиональная реабилитация играет большую роль в разработке программ упражнений и обучении упражнениям на растяжку, чтобы минимизировать ограничивающие контрактуры.Тяжелые контрактуры и сколиоз лечат хирурги-ортопеды из нашей команды, которые имеют большой опыт лечения МДД. Функция сердца отслеживается с помощью ЭКГ и эхокардиограммы, проверенных кардиологом. Эндокринолог в команде занимается гормональными проблемами и здоровьем костей. Центр генетических мышечных заболеваний разработан с целью максимизировать независимую функцию и минимизировать осложнения МДД. Мы приветствуем возможность работать с семьями, живущими с МДД.
Дополнительные ресурсы:
Синдром невуса Беккера у педиатрической пациентки
Синдром невуса Беккера является частью синдромов эпидермального невуса и был описан с фенотипом, который включает невус Беккера, ипсилатеральную гипоплазию молочной железы и различные пороки развития скелета.Это чаще встречается у мужчин, чем у женщин (5: 1), но более актуально у женщин. Диагноз ставится на основании клинических данных, поражение кожи должно присутствовать, и никаких других пронумерованных критериев не установлено, но при наличии большего количества критериев вероятность диагноза выше. Что касается лечения гипоплазии груди, использование антиандрогенных препаратов продемонстрировало адекватный клинический ответ в дозе спиронолактона 50 мг / день.
1. Введение
Невус Беккера — это эпидермальная кожная гамартома, которая чаще всего встречается у подростков и чаще встречается у мужчин, чем у женщин.Синдром невуса Беккера — очень редкий синдром, характеристики которого включают невус Беккера и гипоплазию ипсилатеральной молочной железы, а также скелетные, мышечные или другие кожные заболевания.
2. История болезни
Пациентка 11 лет, ранее перенесшая хирургическую коррекцию пупочной грыжи, обратилась за консультацией по поводу гипоплазии левой груди. При физикальном обследовании выявлено врожденное пигментное поражение кожи размером 3 × 4 см с неровными границами и гипертрихоз в области левой нижней челюсти (рис. 1).В грудной клетке копытце было обнаружено развитие Tanner III-IV правой груди и развитие Tanner II левой груди плюс выраженная гипоплазия (Рисунок 2). Биопсия кожного поражения выявила увеличение количества пучков гладких мышц и волосяных фолликулов, а также увеличенные сосочковые гребни с пигментацией в базальном эпидермисе без признаков злокачественности (рис. 3). Биопсия была совместима с невусом Беккера. Ультразвуковое исследование почек, функция почек, рентгенография грудной клетки и позвоночника были нормальными. Ультразвуковое исследование грудной клетки исключило отсутствие молочных желез.Уровни гормонов (тестостерон, пролактин, эстрадиол, ФСГ, ЛГ и ТТГ) были нормальными. Диагноз синдрома невуса Беккера был поставлен из-за сочетания невуса Беккера, односторонней гипоплазии груди, пупочной грыжи и грыжевой мышцы. Пациентка отреагировала на терапию спиронолактоном значительным улучшением развития левой груди (рис. 4).
3. Обсуждение
Невус Беккера (BN) — эпидермальная кожная гамартома, впервые описанная в 1949 году Беккером [1].Обычная клиническая картина — множественные пятна, связанные с гипертрихозом и гиперпигментацией туловища или плеча с односторонним распределением. Он также может быть представлен как бесшерстный BN. Большинство исследований у взрослых показывают, что заболеваемость у мужчин в пять раз выше, чем у женщин, но несколько других исследований выявили преобладание женщин [2]. Более высокая заболеваемость у мужчин, о которой сообщалось в тематических исследованиях взрослых, не была обнаружена у детей [3]. Гистологическое исследование выявляет акантоз, гиперпигментацию эпидермиса, папилломатоз, гиперплазию арректорных мышц и меланофаги в дерме [4].Несмотря на гиперпигментацию, BN не входит в состав меланоцитарных поражений и считается особой формой эпидермальных невусов.
В прошлом различные авторы связывали БН с другими нарушениями опорно-двигательного аппарата, но в 1997 году Хэппл и Купман описали синдром невуса Беккера (БНС) [5]. BNS — очень редкий синдром, характеристики которого включают невус Беккера и гипоплазию ипсилатеральной молочной железы, а также скелетные, мышечные или другие кожные заболевания. Входит в группу синдромов эпидермальных невусов, с синдромом Протея, синдромом комедонического невуса, пигментокератотическим факоматозом, синдромом сальных невусов, синдромом ДЕТЕЙ [5, 6].Диагноз является клиническим и должен включать наличие невуса Беккера, а также необходимы другие кожные, мышечные и / или скелетные нарушения, которые могут включать ипсилатеральную гипоплазию плеча или руки, ипсилатеральную гипоплазию груди, избыточный сосок, асимметрию лица, гипоплазию кожи. височная область, spina bifida, спондилодез, pectus carinatum или excatum, сколиоз, липодистрофия, стрессовые переломы, добавочная мошонка, контралатеральная гипоплазия малых половых губ и пупочная грыжа [7, 8].Патогенез остается неясным; большинство случаев происходит спорадически, но может произойти знакомое вовлечение из-за неполной пенетрантности аутосомно-доминантного наследования. Андрогены были задействованы из-за более широкого проявления у подростков и мужчин, а также из-за связи с гипертрихозом, акне и аномалиями мошонки [9]. Поскольку андрогены также связаны с развитием костей, мышц и волос, это может играть роль в патогенезе других системных изменений. Поражения BN имеют большее количество рецепторов андрогенов, чем нормальная кожа; это говорит о том, что BNS следует рассматривать как часть спектра синдрома гиперчувствительности к андрогеновым рецепторам.Гипоплазию груди можно объяснить уравновешивающим действием андрогенов, снижающих эффект эстрогенов [10].
Специфического лечения BNS не описано; поскольку поражения BN обычно большие, хирургическое лечение ограничено. Лечение рубиновым лазером с модуляцией добротности (694 нм) и эрбиевым: YAG-лазером дало удовлетворительные результаты. Также при гипертрихозе можно использовать различные успешные техники депиляции [11]. Сообщалось об удовлетворительном ответе со значительным уменьшением гиперпигментации при местном применении флутамида, нестероидного антиандрогена [12].Спиронолактон — это антиандроген, который используется при других дерматологических заболеваниях из-за гормонального действия. Что касается гипоплазии груди, его действие до конца не изучено, но было предложено реагировать на отрицательную обратную связь рецепторов андрогенов. При дозировке 50 мг / день описано улучшение гипоплазии груди [13].
4. Заключение
Случай, представленный 11-летней пациенткой с дерматологическим поражением, клинически и гистологически совместимым с БН, ассоциированным с предыдущей пупочной грыжей, ипсилатеральной гипоплазией груди и открытой грудной клеткой, соответствует критериям, описанным для Беккера. синдром невуса.Пациентка имела отличный ответ на терапию спиронолактоном в отношении объема груди; это согласуется с другими случаями, о которых сообщалось ранее. Литература также поддерживает то, что пациентов с BN следует обследовать на предмет связанных с ней аномалий BNS.
Конкурирующие интересы
Авторы заявляют об отсутствии конфликта интересов в отношении публикации данной статьи.
Авторские права
Авторские права © 2016 Сара Эрнандес-Кисено и др. Это статья в открытом доступе, распространяемая под лицензией Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии правильного цитирования оригинальной работы.
Синдром невуса Беккера нижней части тела: один случай и обзор литературы | HTML
Синдром Беккера-Невуса нижней части тела: новый случай и обзор литературы
Кристина Шефер 1 , Борис Бауэр 1 , Джулиан Донхаузер 2 , Андреас Керстан 1 и Хеннинг Хамм 1
1 Отделение дерматологии, венерологии и аллергологии и 2 Институт диагностической и интервенционной радиологии, Университетская клиника Вюрцбурга, Вюрцбург, Германия
Синдром невуса Беккера — это редкий синдром эпидермального невуса, определяемый по сочетанию невуса Беккера с различными кожными, мышечными и скелетными аномалиями.В большинстве случаев аномалии включают исключительно ипсилатеральную гипоплазию груди, ареолы и / или соска в дополнение к невусу. Здесь мы сообщаем о 42-летней женщине с обширным невусом Беккера, простирающимся от левой ягодицы до левой голени, подтвержденным гистологическим исследованием. Кроме того, наблюдалась выраженная гипоплазия жировой ткани левого бедра, подтвержденная магнитно-резонансной томографией, в отличие от гипоплазии жировой ткани левой ягодичной области. Основные мышцы и кости не были затронуты.Не было разницы в длине ног. Кроме того, мы рассматриваем и обсуждаем особенности синдрома невуса Беккера с акцентом на 10 зарегистрированных случаев с вовлечением нижней части тела.
Ключевые слова : невус Беккера; Синдром невуса Беккера; гипоплазия жировой ткани; нижняя часть тела.
Принята к печати 23 ноября 2016 г .; Epub до печати 24 ноября 2016 г.
Acta Derm Venereol 2017; 97: XX – XX.
Corr: Хеннинг Хамм, отделение дерматологии, венерологии и аллергологии, Университетская клиника Вюрцбурга, Josef-Schneider-Str.2, DE-97080 Вюрцбург, Германия. E-mail: [email protected]
Невус Беккера — довольно распространенная доброкачественная аномалия, клинически характеризующаяся пятном или плоской бляшкой от светло- до темно-коричневого цвета со средним размером 125 см² и неровными границами (1) . В большинстве случаев невус Беккера локализуется односторонне на верхней части туловища, в основном в области плеча, груди или лопатки (2). Из-за стимуляции андрогенами он часто проявляется гипертрихозом и более темной окраской у постпубертатных мужчин. Tymen et al.(1) сообщили о распространенности, близкой к 0,52%, тогда как De Almeida et al. (3) поставили диагноз у 4,19% 18-летних бразильских мужчин. Вероятно, что более выраженная видимость и, как следствие, большая эстетическая неудобства у подростков и взрослых мужчин привели к ошибочному предположению о существенном преобладании мужчин до 4,5: 1 (4).
При гистологическом исследовании невус Беккера показывает гиперпигментацию базального слоя с незначительным увеличением количества меланоцитов или без него, изменчивый акантоз с легким папилломатозом и удлинение ребер сетчатки с плоскими кончиками.Как правило, в дерме обнаруживается гипертрофия пилорум мышц и пучков гладкомышечных волокон, не связанных с волосяными фолликулами (5, 6).
Было описано довольно много дерматозов и кожных опухолей в пространственной связи с невусом Беккера, таких как угри (7–11), дерматит (12), красный плоский лишай (13–15), разноцветный лишай (16–18), кольцевидная гранулема (19), гипогидроз (20), ихтиоз (21), болезнь Боуэна (22), базальноклеточная карцинома (23), лимфоматоидный папулез (24), множественная лейомиома кутиса (25), остеома кутиса (26) и лежащая в основе десмоидная опухоль (27).
В редких случаях невус Беккера также наблюдался в связи с аномалиями развития. Так, в 1997 году Happle & Koopman (28) ввел термин синдром невуса Беккера (BNS) для одновременного присутствия невуса Беккера с определенными кожными и подкожными, мышечными и скелетными дефектами. Основываясь на редких сообщениях о семейном невусе Беккера (29–32) и одном семейном возникновении BNS (33), авторы предложили парадоминантное наследование. Однако генетическая основа и способ передачи до сих пор не выяснены.
В отличие от Becker naevi, соотношение полов в BNS показывает преобладание женщин в опубликованных случаях 1,5: 1 (34). Однако это соотношение может быть неверным в пользу женских случаев, поскольку синдром легче распознается у женщин. Причина в том, что наиболее частая аномалия при BNS состоит из ипсилатеральной гипоплазии груди, ареолы и / или соска, которая гораздо более очевидна у женщин, хотя и не ограничивается ими. Другие связанные признаки включают избыточные соски, сколиоз, позвоночные аномалии и укорочение или другой вид асимметрии верхних конечностей из-за гипоплазии мягких тканей (34).В то время как BNS обычно ограничивается верхней частью тела, наблюдения с участием нижней части тела редки. Мы сообщаем о случае BNS, затрагивающего одну ногу и ягодицу, и рассматриваем 73 ранее опубликованных случая BNS с особым вниманием к его возникновению на нижней части тела.
42-летняя женщина обратилась за косметической консультацией по поводу темно-коричневой гиперпигментации левой ягодичной области и левого бедра. Особенно ее беспокоила кожная складка на левой ягодице, свисавшая над местом прикрепления бедра.Она сообщила, что пигментация присутствовала с рождения и постепенно становилась темнее с начала полового созревания. Усиление гиперпигментации сопровождалось увеличением неровности поверхности кожи и ростом волос в этой области. Кроме того, пациентка отметила медленное увеличение объема левой ягодичной области и уменьшение окружности левого бедра в течение последних 5 лет. В остальном ее история болезни ничем не примечательна. Подобных случаев в ее семье не было.
При физическом осмотре были обнаружены агрегированные фолликулярные слегка приподнятые коричневатые папулы, сливающиеся с пигментными бляшками в левой ягодичной области, дорсальной части левого бедра и проксимальном отделе голени ( Рис. 1 ). Гипертрихоза не было, так как ноги были чисто выбриты. Однако дерматоскопия показала коротко остриженные терминальные волоски. Окружность ее левого бедра была значительно уменьшена по сравнению с противоположной стороной (52,5 против 57,5 см).
Рис.1.Синдром невуса Беккера, затрагивающий левую ягодицу и ногу. Обратите внимание на гиперплазию жировой ткани левой ягодичной области в отличие от гипоплазии жировой ткани бедра. (А) Общий вид. (B) Вид сбоку левой ягодицы и вставка бедра. (C) Деталь нижней части бедра и впадины колена.
Кроме того, жировая ткань левой ягодицы была более объемной, чем правой (расстояние от подвздошной кости до прикрепления бедра 41 против 39 см). Ноги были одинаковой длины.Биопсия тыльной стороны бедра показала эпидермальный акантоз и гиперпигментацию базального слоя без явного увеличения меланоцитов, а также гипертрофию гладкомышечных волокон, характерную для невуса Беккера ( рис. 2 ). Магнитно-резонансная томография таза и ног подтвердила гипоплазию жировой ткани левого бедра и небольшую гиперплазию жировой ткани левой ягодичной области без поражения опорно-двигательного аппарата ( рис. 3 ).
Рис. 2. Гистология левого бедра показывает небольшой папилломатоз, акантоз и гиперпигментацию базального слоя эпидермиса в сочетании с аномальными и разделенными пучками гладких мышц в ретикулярной дерме (гематоксилин и эозин, первоначальное увеличение × 100).
Рис. 3. Магнитно-резонансная томография бедра и левой ягодицы. (A) Магнитно-резонансная томография бедренных костей, показывающая гипоплазию жировой ткани на левом бедре, а также утолщенную и мягко усиливающуюся кутику, соответствующую клиническим данным (изображение T1 Turbo spin echo [TSE], насыщенное жиром после введения i.v. Контраст Gd-DO3A-бутрол, осевой вид). Комментарий: Жировая ткань выглядит темной, контрастные структуры (сосуды, кутис) — яркими. Мышцы показывают промежуточную интенсивность сигнала. (B) Магнитно-резонансная томография, показывающая заметную жировую ткань на левой ягодице и гипоплазию жировой ткани на дорсальной стороне проксимального отдела левого бедра по сравнению с вентральной областью (изображение T1 TSE до введения внутривенного контраста, сагиттальный вид). Комментарий: Жировая ткань, включая костный мозг, выглядит яркой.Мышцы показывают интенсивность сигнала от низкой до средней.
Дальнейшее обследование всего тела не выявило отклонений. В частности, рост груди, подмышечных и лобковых волос, а также опорно-двигательный аппарат верхней части тела не отличался.
BNS обозначает невус Беккера в сочетании с другими кожными и внекожными аномалиями. Возможные ассоциации были хорошо задокументированы в всеобъемлющем обзоре 55 случаев BNS, опубликованном до 2004 г. (34). Всего на сегодняшний день зарегистрировано 73 случая. Таблица I содержит сводку всех наблюдаемых аномалий (1, 4, 6, 33, 35–79). Следует отметить, что невус Беккера на лице является непостоянным признаком челюстно-лицевого синдрома с поражением костей, зубов, десен и кожи, который известен под названиями гемимаксилло-лицевой дисплазии (80, 81), сегментарной одонтомаксиллярной дисплазии (82–84) и HATS ( синдром увеличения челюстно-лицевой области, асимметрии лица, аномалий зубов и кожных покровов) (85, 86). Из-за неясной связи с BNS случаи синдрома HATS не рассматривались в таблице I.
Таблица I. Внекожные аномалии при синдроме невуса Беккера, согласно литературным данным
Кроме того, сомнительный случай BNS с предполагаемым невусом Беккера с двух сторон с поражением генитально-грудной области и шеи, связанный с исключительными неврологическими, эндокринными, сердечными и другими аномалиями. , не был включен (87).
Помимо кожи, молочной железы и жировой ткани, в BNS преимущественно вовлечены кости и мышцы. Обычно ассоциированные аномалии обнаруживаются ипсилатерально по отношению к невусу.Только в 2 случаях поражалась контралатеральная сторона тела (45, 62), а двустороннее поражение было отмечено только в одном случае (41). За исключением 3 случаев, все связанные аномалии носили гипопластический характер, в первую очередь затрагивая женскую грудь. Примечательно, что различие между жировой тканью и мышечной гипоплазией было проведено лишь в небольшом количестве случаев. Пункты гиперплазии включали отек мягких тканей ипсилатеральной кисти (70), гиперплазию ипсилатеральной нижней конечности (72) и увеличение ипсилатеральной стопы (71).В представленном нами случае отмечена гипертрофия подкожно-жировой клетчатки ягодичной области ипсилатеральной к невусу.
Как и в несиндромальных случаях, наиболее частыми участками невуса Беккера в BNS были плечо, передняя часть грудной клетки и лопаточная область. Реже невус располагался в нижней части тела. Lucky et al. были первыми, кто описал пациента с БНС нижних конечностей в 1981 г. (71). Включая наш отчет, до сих пор в англоязычных публикациях было сообщено о 10 случаях поражения БНС на частях тела ниже талии (, таблица II, ).Семь пациентов были женского пола и трое — мужского пола, средний и средний возраст составлял 30 и 34 года соответственно.
Таблица II. Опубликованные случаи синдрома невуса Беккера с вовлечением нижней части тела
Помимо нашего случая, зарегистрированы еще 3 пациента с BNS с липоатрофией ипсилатеральной ноги (48, 73), а в другом случае наблюдалась гипертрофия ипсилатеральной нижней конечности ( 72). В 2 случаях была укорочена нога, пораженная невусом (47, 69). Другие аномалии включали двусторонний внутренний перекрут большеберцовой кости (41), добавочную промежность мошонки (76) и гипоплазию контралатеральной губы минус (45).У 4 пациентов с невусом Беккера на нижней конечности ассоциированная аномалия локализовалась в верхней части тела (59, 60, 63, 78).
Действительно ли BNS поражает нижнюю часть тела значительно реже, чем верхнюю, или же она недооценивается в этой области — вопрос предположений. Из-за преимущественного расположения невуса Беккера на верхней части туловища, а также из-за пространственного отношения к внекожным аномалиям, есть соблазн предположить реальную разницу в частоте поражения участков тела.С другой стороны, когда он ограничен нижней частью тела и не имеет признаков односторонней гипоплазии груди, диагностика BNS является более сложной задачей. Ряд случаев BNS, локализованных в необычных местах, могли быть ошибочно приняты за другие формы меланоза или за врожденные меланоцитарные невусы (CMN).
Несколько фенотипов кожной мозаики, кроме невуса Беккера, могут встречаться в сочетании с врожденной гипоплазией или, реже, гиперплазией подкожно-жировой клетчатки и более глубоких структур. Уменьшение объема подкожной ткани — хорошо известная особенность больших и гигантских ЦМН, которая недавно была подчеркнута в исследовании, посвященном вопросам профилирования роста и гормонов (88).Также регистрируются липоматозные образования мягких тканей при ЦМН с диффузной инфильтрацией невомеланоцитов в жировые дольки (89). Большинство крупных, гигантских и множественных ЦМН вызываются онкогенными мутациями в кодоне 61 гена NRAS (88). Асимметрия тела, возникающая в результате односторонней гипо- или гиперплазии туловища и / или конечностей, является наиболее частой внекожной аномалией, связанной с врожденной телеангиэктатической кожной оболочкой (90). Аналогичным образом, выраженная липогипоплазия правой ягодицы и бедра, а также гипоплазия груди наблюдались при цезиофламмеа факоматозе (91).Недавно было показано, что этот и другие типы пигментно-сосудистого факоматоза принадлежат к группе мозаичных гетеротримерных нарушений G-белков путем обнаружения активирующих мутаций в генах GNA11 и GNAQ (92). Нарушение регуляции жировой ткани, включая липомы, липогипоплазию, избыточный жировой рост и локализованные жировые отложения, является частым проявлением синдрома Протея, вызванного соматическими активирующими мутациями в гене, кодирующем стимулирующую рост серин / треонинкиназу AKT1 (93).Аналогичным образом, связанные расстройства избыточного роста со значительным клиническим перекрытием друг друга, такие как избыточный рост фиброзно-жировой ткани, множественный липоматоз с гемигиперплазией, врожденный избыточный липоматозный рост, сосудистые мальформации, эпидермальный невус, сколиоз / скелетно-спинномозговой синдром (КЛОВ) и мегалэнцефалия-капиллярная мальформация. синдрома, вызваны мутациями в других сигнальных белках пути RTK / PI3K / AKT / mTOR и постоянно присутствуют с нарушением регуляции жировой ткани, будь то липоматозные поражения или региональная липогипоплазия (94).Мозаицизм летальных аутосомных мутаций, совместимых только с жизнью в мозаичном состоянии, был доказан на молекулярном уровне для синдромов Proteus, CLOVES и MCM, а также для больших / гигантских CMN (95).
Напротив, молекулярная основа BNS неизвестна. Основываясь на спектре внекожных аномалий, можно предположить, что причинный ген (ы) участвует в регуляции распределения жировой ткани. Поскольку BNS экспрессируется сегментарно, летальная аутосомная мутация, сохранившаяся только в виде мозаики, кажется наиболее вероятным генетическим объяснением.
Хотя пациенты могут чувствовать значительное беспокойство из-за заметного невуса Беккера, терапевтические возможности ограничены. Последствия хирургического удаления могут быть более заметными, чем исходное поражение. Удаление пигмента с помощью лазерной абляции Er: YAG оказалось более эффективным, чем лечение лазером Nd: YAG с модуляцией добротности (96). Абляционная фракционная лазерная терапия была оценена как умеренно эффективная, но результаты были ухудшены поствоспалительной гиперпигментацией (97). Один случай был успешно вылечен местным флутамидом (98).Спиронолактон в качестве антиандрогенного агента был опробован на одной женщине с гипоплазией груди и, как сообщалось, привел к ее увеличению через месяц после начала терапии (55). В некоторых случаях гипоплазия груди и аномалии костей, такие как сколиоз, корректировались хирургическим путем.
В заключение, невус Беккера является довольно распространенной доброкачественной аномалией, тогда как его связь со структурными аномалиями мягких тканей, мышц и костей — редкое явление. С момента первоначального описания 73 наблюдения BNS, за исключением сомнительных случаев на лице, были опубликованы в медицинской литературе, которую можно найти в PubMed / MEDLINE.С учетом нашего отчета нижняя часть тела была задействована в 10 случаях BNS. Пациенты с диагнозом невус Беккера должны пройти клиническое обследование на предмет связанных мягких тканей, мышечных и скелетных аномалий. При подозрении на BNS предпочтительным методом визуализации является магнитно-резонансная томография, которая может подтвердить диагноз и рекомендуется перед любым хирургическим вмешательством. Варианты лечения ограничены.
Авторы благодарны Герхарду А. за отличные клинические фотографии.Кремер, отделение дерматологии, венерологии и аллергологии, Университетская клиника Вюрцбурга, Германия.
Авторы заявляют об отсутствии конфликта интересов.
- Tymen R, Forestier JF, Boutet B, Colomb D. Невус позднего Беккера. Сто дел (пер. Автора). Ann Dermatol Venereol 1981; 108: 41–46.
Просмотреть статью Google Scholar - Альфадли А., Хайнау Б., Аль-Робаи А., Банка Н. Меланоз Беккера: отчет о 12 случаях с атипичными проявлениями.Int J Dermatol 2005; 44: 20–24.
См. Статью Google Scholar - Де Алмейда Х.Л., Дукиа Р.П., Соуза П.Р., Бреуниг Жде А. Распространенность и характеристики невуса Беккера у бразильских 18-летних мужчин. Int J Dermatol 2010; 49: 718–720.
Просмотреть статью Google Scholar - Ламберт Дж. Р., Виллемс П., Абс Р., Ван Рой Б. Невус Беккера, связанный с хромосомным мозаицизмом и врожденной гиперплазией надпочечников. J Am Acad Dermatol 1994; 30: 655–657.
Просмотреть статью Google Scholar - Ким Й.Дж., Хан Дж.Х., Кан Х.Й., Ли Э.С., Ким Ю.Сверхэкспрессия рецепторов андрогенов в невусе Беккера: гистопатологический и иммуногистохимический анализ. Дж. Кутан Патол, 2008 г .; 35: 1121–1126.
Просмотреть статью Google Scholar - Альфаро А., Торрело А., Эрнандес А., Замбрано А., Хаппл Р. Синдром невуса Беккера. Actas Dermosifilogr 2007; 98: 702–704.
Просмотреть статью Google Scholar - Burgreen BC, Ackerman AB. Акнеформные поражения при невусе Беккера. Cutis 1978; 21: 7617–7619.
Просмотреть статью Google Scholar - Agrawal S, Garg VK, Sah SP, Agarwalla A.Акне при невусе Беккера. Int J Dermatol 2001; 40: 583–585.
Просмотреть статью Google Scholar - Person JR, Longcope C. Невус Беккера: андроген-опосредованная гиперплазия с повышенными рецепторами андрогенов. J Am Acad Dermatol 1984; 10: 235–238.
См. Статью Google Scholar - Даунс AM, Mehta R, Lear JT, Peachey RD. Акне в невусе Беккера: связь, опосредованная андрогенами? Clin Exp Dermatol 1998; 23: 191–192.
Просмотреть статью Google Scholar - Juhl M, Pappo E, Bain M.Акне, выделенное в невусе Беккера у 14-летней девочки. Dermatol Online J 2015, 15 августа; 21 (8).
Просмотреть статью Google Scholar - Локшин Б.Н., Спринг С., Броган Б., Биллингс С. Экзематозный дерматит и узловатая пруриго ограничиваются невусом Беккера. Int J Dermatol 2006; 45: 1465–1466.
См. Статью Google Scholar - Terheyden P, Hornschuh B, Karl S, Becker JC, Bröcker EB. Красный плоский лишай, связанный с невусом Беккера. J Am Acad Dermatol 1998; 38: 770–772.
Просмотреть статью Google Scholar - Puri S, Nanda S, Grover C, Batra VV, Garg VK, Reddy BS. Врожденный невус Беккера с красным плоским лишаем. Педиатр Дерматол 2005; 22: 328–330.
Просмотреть статью Google Scholar - Гупта С. Гупта С., Аггарвал К., Джайн В.К. Невус Беккера с витилиго и красным плоским лишаем: коктейль дерматозов. N Am J Med Sci 2010; 2: 333–335.
См. Статью Google Scholar - Wright RC. Еще одна ассоциация с невусом Беккера. Arch Dermatol 1979; 115: 1035.
Просмотреть статью Google Scholar - Сингал А., Бхаттачарья С. Н., Кумар С., Баруа М. С.. Разноцветный гипопигментный лишай на невусе Беккера: надежда на новый метод лечения? Indian J Dermatol Venereol Leprol 1998; 64: 137–138.
Просмотреть статью Google Scholar - Bhogal CS, Singal A, Baruah MC. Разноцветный гипопигментный лишай, развивающийся на ранее существовавшем невусе Беккера. Индийский J Dermatol Venereol Leprol 2002; 68: 43–44.
См. Статью Google Scholar - Weinberg JM, Scheinfeld N, Tishler HR.Кольцевидная гранулема ограничена невусом Беккера. Br J Dermatol 2004; 151: 245–246.
Просмотреть статью Google Scholar - Do JE, Kim YJ, Kang HY. Гипогидроз совмещен с невусом Беккера. Br J Dermatol 2007; 156: 766–767.
См. Статью Google Scholar - Criscione V, Telang GHT. Невус Беккера с ихтиотическими особенностями. Arch Dermatol 2010; 146: 575–577.
Просмотреть статью Google Scholar - Honda M, Suzuki T, Kudoh K, Tagami H. Болезнь Боуэна, развивающаяся при меланозе Беккера (невус Беккера).Br J Dermatol 1997; 137: 659–661.
Просмотреть статью Google Scholar - Patrizi A, Medri M, Neri I, Fanti PA. Невус Беккера ассоциирован с базальноклеточным раком, меланоцитарным невусом и гладкомышечной гамартомой. J Eur Acad Dermatol Venereol 2007; 21: 130–132.
Просмотреть статью Google Scholar - Buder K, Wendel AM, Cerroni L, Goebeler M, Kerstan A. Случай лимфоматоидного папулеза, ограниченный меланозом Беккера. Дерматология 2013; 226: 224–227.
Просмотреть статью Google Scholar - Thappa DM, Garg BR, Prasad RR, Ratnakar C.Множественная кожная лейомиома, связанная с невусом Беккера. J Dermatol 1996; 23: 719–720.
Просмотреть статью Google Scholar - Park SBM, Song BH, Park EJ, Kwon ICH, Kim KH, Kim KJ. Случай невуса Беккера с кожной остеомой. Энн Дерматол 2011; 23: 247–249.
См. Статью Google Scholar - Sciallis GF, Sciallis AP. Невус Беккера с лежащей в основе десмоидной опухолью: описание случая и обзор, включая опыт клиники Мэйо. Arch Dermatol 2010; 146: 1408–1412.
См. Статью Google Scholar - Happle R, Koopman RJ.Синдром невуса Беккера. Am J Med Genet 1997; 68: 357–361.
Просмотреть статью Google Scholar - Фретцин Д.Ф., Уитни Д. Семейный невус Беккера. J Am Acad Dermatol 1985; 12: 589–590.
См. Статью Google Scholar - Jain HC, Fisher BK. Семейный невус Беккера. Int J Dermatol 1989; 8: 263–264.
См. Статью Google Scholar - Panizzon RG. Семейный невус Беккера. Int J Dermatol 1990; 29: 158.
Просмотреть статью Google Scholar - Book SE, Glass AT, Laude TA.Врожденный невус Беккера с семейной ассоциацией. Pediatr Dermatol 1997; 14: 373–375.
Просмотреть статью Google Scholar - Maessen-Visch MB, Hulsmans RFHJ, Hulsmans FJH, Neumann HAM. Невиформный меланоз Беккера и сколиоз: совпадение? Acta Derm Venereol 1997; 77: 135–136.
Просмотреть статью Google Scholar - Данарти Р., Кёниг А., Салхи А., Биттар М., Хэппл Р. Синдром невуса Беккера. J Am Acad Dermatol 2004; 51: 965–969.
Просмотреть статью Google Scholar - Wosyka H.Множественная лейомиобластома кутиса в сегменте Anordnung. Dermatol Wochenschr 1934; 34: 1110–1112.
Просмотреть статью Google Scholar - Copeman PW, Wilson-Jones E. Пигментированный волосатый эпидермальный невус (Беккер). Arch Dermatol 1965; 92: 249–251.
Просмотреть статью Google Scholar - Mascaro JM, Galy de Mascaro C, Pinol Aguade J. Historia natural del nevus de Becker. Med Cutan Ibero Latinoamer 1970; 4: 437–445.
Просмотреть статью Google Scholar - Naranjo R, Delgado V, de Dulanto F, Herrera E, Armijo M.Меланозис де Беккер. Actas Dermosifiliogr 1980; 71: 331–336.
Просмотреть статью Google Scholar - Smolle J. Becker’sche Melanose und oberflächliche zirkumskripte Sklerodermie. Akt Dermatol 1983; 9: 187–188.
См. Статью Google Scholar - Glinick SE, Alper JC, Bogaars H, Brown JA. Меланоз Беккера: сопутствующие аномалии. J Am Acad Dermatol 1983; 9: 509–514.
См. Статью Google Scholar - Moore JA, Schosser RH. Меланоз Беккера и гипоплазия груди и большой грудной мышцы.Pediatr Dermatol 1985; 3: 34–37.
Просмотреть статью Google Scholar - Friedel J, Champy M, Seltan A, Grosshans E. Что это за диагностика? Невус де Беккера имеет ипсилатеральную гипоплазию груди. Ann Dermatol Venereol 1988; 115: 1059–1060.
Просмотреть статью Google Scholar - Blanc F, Jeanmougin M, Civatte J. Naevus de Becker et hypoplasia mammaire. Ann Dermatol Venereol 1988; 115: 1127.
Просмотреть статью Google Scholar - Formigon M, Alsina MM, Mascaro JM, Rivera F.Невус Беккера и ипсилатеральная гипоплазия молочной железы: исследование рецепторов андрогенов у двух пациентов. Arch Dermatol 1992; 128: 992–993.
Просмотреть статью Google Scholar - Ван Гервен Х. Дж., Купман Р. Дж., Стейлен П. М., Меланоз Хэппла Р. Беккера с локальной липоатрофией и ипсилатеральной гипоплазией груди. Br J Dermatol 1993; 129: 123.
Просмотреть статью Google Scholar - Cambiaghi S, Brusasco A, Tadini G, Alessi E. Nevo di Becker congenito con ipoplasia mammaria ipsilaterale.G Ital Dermatol Venereol 1994; 129: 169–172.
См. Статью Google Scholar - Crone AM, James MP. Гигантский невус Беккера с ипсилатеральной ареолярной гипоплазией и асимметрией конечностей. Clin Exp Dermatol 1997; 22: 240–241.
Просмотреть статью Google Scholar - Ибрагим К., Таллаб Т., Бахамдан К., Кхаре А. Невус Беккера и ипсилатеральная гипоплазия груди. J Saudi Soc Dermatol Venereol 1997; 5: 222–223.
Просмотреть статью Google Scholar - Domján K, Török L.[Синдром Беккера-невуса.] B? Rgyógy Venereol Sz 1999; 75: 3–5 (на венгерском).
Просмотреть статью Google Scholar - Шарма Р., Мишра А. Беккер невус с ипсилатеральной ареолярной гипоплазией у трех мужчин. Br J Dermatol 1997; 136: 471–472.
Просмотреть статью Google Scholar - Биттар М., Дьерна А., Мартинес де Лисардуй И. Синдром невус де Беккера. Буэнос-Айрес: Конгресс Общества дерматологии Аргентины; 1998 г.
Просмотреть статью Google Scholar - Salhi A, Ammar-Khodja A, Grossjans E, Ait-Belkacem F, Heid E, Happle R.Невус де Беккер и синдром невуса де Беккера: 6 cas. Представлено на 6-м Магрибском конгрессе дерматологов; 6–7 октября 1999 г .; Алжир, Алжир.
Просмотреть статью Google Scholar - Анджело К., Гроссо М.Г., Стелла П., Де Сио С., Пассарелли Ф., Пудду П., Парадизи М. Синдром невуса Беккера. Cutis 2001; 68: 123–124.
Просмотреть статью Google Scholar - Сантос-Хуанес Дж., Галаче С., Курто Дж. Р., Карраско М. П., Рибас А., Санчес-дель-Рио Дж. Угри в невусе Беккера и гипоплазия груди.Int J Dermatol 2002; 41: 699–700.
См. Статью Google Scholar - Jung JH, Kim YC, Park HJ, Cinn YW. Невус Беккера с ипсилатеральной гипоплазией груди: улучшение после приема спиронолактона. J Dermatol 2003; 30: 154–156.
Просмотреть статью Google Scholar - Да Ле Торре C, Родригес Гранадос Т., Розон Э, Лосада А. Синдром невуса де Беккера с генерализованными поражениями. Actas Dermosifiliogr 2003; 94: 51.
Просмотреть статью Google Scholar - Раси А, Тагизаде А, Ягмаи Б., Сетарех-Шенас Р.Невус Беккера с ипсилатеральной гипоплазией груди: описание случая и обзор литературы. Arch Iran Med 2006; 9: 68–71.
Просмотреть статью Google Scholar - Sirka CS, Puhan MR, Behera S, Mohanty P, Nevus Нанды М. Беккера с ипсилатеральной гипоплазией груди. Индийский J Dermatol Venereol Leprol 2009; 75: 202–203.
Просмотреть статью Google Scholar - Cosendey FE, Bernhard GA, Azulay DR, Martinez NS, Dias MFRG. Синдром невуса Беккера. Бюстгальтеры Dermatol 2010; 85: 380–384.
Просмотреть статью Google Scholar - Baeta, IGR, Perera ACF, Bittencourt FV, Viotti CV, Da Costa Junior SR. Синдром невуса Беккера — история болезни. Бюстгальтеры Dermatol 2010; 85: 713–716.
Просмотреть статью Google Scholar - Фернандес С., Агравал А., Шрешта Б. Б., Йоги Н., Чериан Л. Синдром невуса Беккера с квадрипарезом. BMJ Case Reports 2010; DOI: 10.1136 / bcr.6.2010.3117.
Просмотреть статью Google Scholar - Patrizi A, Medri M, Raone B, Bianchi F, Aprile S, Neri I.Клиническая характеристика невуса Беккера у детей. Отчет о 118 случаях из Италии. Pediatr Dermatol 2012; 29: 571–574.
Просмотреть статью Google Scholar - Намкун С., Ким Дж. Й., Гай Дж, Чунг Дж., Хонг С. П., Парк Британской Колумбии, Ким М. Х. Пигментная эпителиоидная меланоцитома возникла у пациента с синдромом невуса Беккера. J Dermatol 2012; 39: 811–812.
Просмотреть статью Google Scholar - Schneider ME, Drahts R, Fritsche E, Hug U. Интересный случай: односторонняя гипоплазия груди при синдроме невуса Беккера.Хандчир Микрочир Пласт Чир 2014; 46: 266–267.
Просмотреть статью Google Scholar - Пектас С.Д., Акоглу Г., Метин А., Адияман Н.С., Демирсерен М.Э. Синдром невуса Беккера проявляется ипсилатеральной гипоплазией груди. Индийский J Dermatol 2014; 59: 634.
Просмотреть статью Google Scholar - Урбани К.Э., Бетти Р. Сверхштатные соски и невус Беккера: ранее не описываемая ассоциация. Отчет о 9 пациентах, включая подгруппу с уропатиями. Eur J Dermatol 1995; 5: 685–687.
Просмотреть статью Google Scholar - Урбани К.Э., Бетти Р. Дополнительные соски, встречающиеся вместе с невусом Беккера: ассоциация, включающая одну общую парадоминантную черту? Hum Genet 1997; 100: 388–390.
Просмотреть статью Google Scholar - Урбани CE, Бетти Р. Полителия в невусе Беккера. Дерматология 1998; 196: 251–252.
Просмотреть статью Google Scholar - Сарате-Мойсен А., Ферегрино-Эрнандес Х.Э., Дельгадо Фернандес А., Вега-Мернихе Э. Меланозис де Беккер: информация о казино.Bol Med Hosp Infant Mex 1992; 49: 762–765.
Просмотреть статью Google Scholar - Верма С., Шарма Ю.К., Део К., Гупта А. Синдром невуса Беккера; вероятно, первое сообщение о сопутствующем акрально локализованном врожденном BN, гипертрофии мягких тканей и сращении костей запястья. J Eur Acad Dermatol Venereol 2016; 30: 184–186.
См. Статью Google Scholar - Lucky AW, Saruk M, Lerner AB. Невус Беккера связан с асимметрией конечностей. Arch Dermatol 1981; 117: 234.
См. Статью Google Scholar - Чанг Х.М., Чанг Ю.Т., Чен С.Л., Ван В.Дж., Вонг К.К.Меланоз Беккера, связанный с ипсилатеральной гиперплазией нижних конечностей и pectus excatum: отчет о болезни и обзор литературы. Dermatol Sinica 2002; 20: 27–32.
Просмотреть статью Google Scholar - Cox NH. Невус бедра Беккера с липоатрофией. Отчет о двух случаях. Clin Exp Dermatol 2002; 27: 27–28.
См. Статью Google Scholar - Gauglitz G, Müller DS, Molin S, Ruzicka T, Herzinger T. Невус Беккера на ноге с липоатрофией. JAMA Dermatol 2013; 149: 1115–1116.
Просмотреть статью Google Scholar - Рамот Ю., Малый А., Злотогорский А. Редкий случай множественных невусов Беккера в мозаичном узоре шахматной доски. J Eur Acad Dermatol Venereol 2014; 28: 1573–1574.
Просмотреть статью Google Scholar - Силит Дж. А., Гроссман М. Е., Луйандо Й, Оларте М. Р., Невус Наглера Х. Беккера и дополнительная мошонка: уникальное явление. J Am Acad Dermatol 1986; 14: 905–907.
Просмотреть статью Google Scholar - Hulsmans FJH, Hulsmans RFHJ, Dijkstra PF, van Ooy A.Патологии мягких тканей и костей, связанные с невиформным меланозом Беккера: клинико-радиологическое исследование с участием 40 пациентов. Br J Dermatol 1989; 121: 524–526.
Просмотреть статью Google Scholar - Steiner D, Pessanha ACAF, Feola C, de Norais e Silva FA, Bialeski N, Buzzoni CAB. Вы знаете этот синдром? Бюстгальтеры Dermatol 2011; 86: 165–166.
См. Статью Google Scholar - Glinick SE, Alper JA. Спектр изменений меланоза Беккера больше, чем предполагалось.Arch Dermatol 1986; 122: 375.
Просмотреть статью Google Scholar - Friedlander-Barenboim S, Kamburo? Lu K, Kaffe I. Клинические и радиологические проявления гемимаксилло-лицевой дисплазии / сегментарной одонто-челюстной дисплазии: критический анализ и отчет о случае. Oral Surg Oral Med Oral Pathol Oral Radiol 2012; 113: 268–273.
Просмотреть статью Google Scholar - Kim HJ, Kim KD, Lee MH. Меланоз Беккера, связанный с фиброзной дисплазией. Int J Dermatol 2002; 41: 284–286.
Просмотреть статью Google Scholar - Petraud A, Sury F, Perrinaud A, de Pinieux G, Laure B, Goga D. Невус Беккера и гемимаксиллярная гипоплазия: совпадение или синдром? Rev Stomatol Chir Maxillofac Chir Orale 2013; 114: 34–37.
См. Статью Google Scholar - Jones AC, Ford MJ. Одновременное возникновение сегментарной одонтомаксиллярной дисплазии и невуса Беккера. J Oral Maxillofac Surg 1999; 57: 1251–1254.
Просмотреть статью Google Scholar - Рай С., Малик Р., Панджвани С., Мисра Д., Верма С.Односторонняя сегментарная одонтомаксиллярная дисплазия: редкая форма из 3 случаев и обзор. Индийский J Dent Res 2014; 25: 102–106.
Просмотреть статью Google Scholar - Alshaiji JM, Handler MZ, Huo R, Freedman A, Schachner LA. Синдром HATS: увеличение гемимаксилля, асимметрия лица, аномалии зубов и кожных покровов. Cutis 2014; 94: E18–21.
См. Статью Google Scholar - Welsch MJ, Stein SL. Синдром увеличения гемимаксилляров, асимметрии лица, аномалий зубов и кожных проявлений (HATS).Pediatr Dermatol 2004; 21: 448–451.
Просмотреть статью Google Scholar - Dasegowda SB, Basavaraj G, Nischal K, Swaroop M, Umashankar N, Swamy SS. Синдром невуса Беккера. Индийский J Dermatol 2014; 59: 421.
Просмотреть статью Google Scholar - Вэлчли Р., Уильямс Дж., Коул Т., Даттани М., Хиндмарш П., Кеннеди Н. и др. Рост и гормональный профиль у детей с врожденными меланоцитарными невусами. Br J Dermatol 2015; 173: 1471–1478.
Просмотреть статью Google Scholar - Патель К.Р., Чернок Р., Льюис Дж. С. мл., Раптис, Калифорния, Аль-Гилани М., Денер Л.П.Липоматозный врожденный меланоцитарный невус, проявляющийся у молодого взрослого в виде образования на шее. Head Neck Pathol 2013; 7: 404–408.
См. Статью Google Scholar - Kienast AK, Hoeger PH. Cutis marmorata telangiectatica congenita: проспективное исследование 27 случаев и обзор литературы с предложением диагностических критериев. Clin Exp Dermatol 2009; 34: 319–323.
Просмотреть статью Google Scholar - Castori M, Rinaldi R, Angelo C, Zambruno G, Grammatico P, Happle R. Phacomatosis cesioflammea с односторонней липогипоплазией.Am J Med Genet A 2008; 146A: 492–495.
Просмотреть статью Google Scholar - Thomas AC, Zeng Z, Rivière JB, O’Shaughnessy R, Al-Olabi L, St-Onge J, et al. Мутации, активирующие мозаику в GNA11 и GNAQ, связаны с пигментно-васкулярным факоматозом и обширным кожным меланоцитозом. J Invest Dermatol 2016; 136: 770–778.
Просмотреть статью Google Scholar - Обзор синдрома Коэна М. М. Младшего Протея: молекулярные, клинические и патологические особенности. Clin Genet 2014; 85: 111–119.
Просмотреть статью Google Scholar - Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, Parker VE, Blumhorst C, Darling T, et al. Клиническое описание и естественный анамнез спектра избыточного роста, связанного с PIK3CA. Am J Med Genet A 2014; 164A: 1713–1733.
Просмотреть статью Google Scholar - Хэппл Р. Категории кожного мозаицизма: предлагаемая классификация. Am J Med Genet A 2016; 170A: 452–459.
Просмотреть статью Google Scholar - Trelles MA, Allones I, Moreno-Arias GA, Vélez M.Невус Беккера: сравнительное исследование эрбия: YAG и неодима с модуляцией добротности: YAG; клинические и гистопатологические данные. Br J Dermatol 2005; 152: 308–313.
Просмотреть статью Google Scholar - Meesters AA, Wind BS, Kroon MW, Wolkerstorfer A, van der Veen JP, Nieuweboer-Krobotová L, et al. Абляционная фракционная лазерная терапия как лечение невуса Беккера: рандомизированное контролируемое пилотное исследование. J Am Acad Dermatol 2011; 65: 1173–1179.
Просмотреть статью Google Scholar - Taheri A, Mansoori P, Sandoval LF, Feldman SR.Лечение невуса Беккера местным флутамидом. J Am Acad Dermatol 2013; 69: e147–148.
Просмотреть статью Google Scholar
Симптомы мышечной дистрофии для типов Дюшенна, Беккера и миотоников
Дистрофия — это любое состояние, при котором часть тела ослабевает или истощается. При мышечной дистрофии слабость проявляется в мышцах. Унаследованная генетическая ошибка не позволяет организму вырабатывать белок, который помогает наращивать мышцы и сохранять их сильными. Только мужчины могут получить определенные типы мышечной дистрофии из-за того, как это заболевание передается по наследству.
Дети, рожденные с мышечной дистрофией, обычно нормально развиваются в течение первых нескольких лет жизни. Они могут внезапно проявить признаки неуклюжести. Эти признаки включают:
- проблемы с ходьбой
- трудности с поднятием передней части стопы (так называемое опускание стопы)
- падение
Со временем дети с мышечной дистрофией могут становиться все слабее и слабее, теряя способность сидеть, ходить, и поднимать предметы. Поскольку болезнь также может поражать мышцы сердца и легких, могут возникнуть одышка и нарушение сердечного ритма.
Существует несколько различных типов мышечной дистрофии. Слабость мышц — отличительный признак каждого типа. Но симптомы могут различаться и проявляться в разном возрасте.
Некоторые мышечные дистрофии легкие. Другие более серьезны и вызывают опасную для жизни мышечную слабость.
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна — наиболее частая и тяжелая форма заболевания. Обычно она начинается в возрасте от 2 до 5 лет.
Симптомы мышечной дистрофии Дюшенна включают:
- Слабость мышц, которая начинается в бедрах, тазе и ногах
- Затруднения при стоянии
- Проблемы с обучением самостоятельно сидеть и ходить
- Неустойчивая походка, ковыляющая
- Ходьба на пальцах ног или подушечках стопы
- Неуклюжесть, частое падение
- Проблемы с подъемом по лестнице
- Затруднения при подъеме из положения лежа или сидя
- Икры большего размера, чем обычно, иногда вызывают боль
- Проблемы с дыханием
- Нарушения обучаемости или поведенческие проблемы
- Искривление позвоночника (сколиоз).Это может привести к тому, что одно бедро поднимется выше другого.
- Проблемы с дыханием, которые в конечном итоге могут потребовать использования аппарата ИВЛ.
К 12 годам большинство детей с мышечной дистрофией Дюшенна должны передвигаться в инвалидном кресле. Болезнь также повреждает сердце и мышцы, необходимые для дыхания, что может быть опасно для жизни.
Мышечная дистрофия Беккера
Симптомы мышечной дистрофии Беккера аналогичны симптомам мышечной дистрофии Дюшенна.Но мышечная дистрофия Беккера начинается позже — примерно в подростковом возрасте. Также он развивается намного медленнее.
Первыми признаками мышечной дистрофии Беккера могут быть проблемы с быстрой ходьбой, бегом и подъемом по лестнице. Другие симптомы могут включать:
- Слабость мышц, которая начинается в тазе, плечах, бедрах и бедрах
- Перевязанная походка
- Ходьба на пальцах ног
- Икры больше, чем обычно
- Мышечные судороги при выполнении упражнений
- Проблемы с подъемом предметы выше уровня талии из-за слабости плеч и рук
- Проблемы с сердцем и дыханием (в более зрелом возрасте)
Часто дети с мышечной дистрофией Беккера могут ходить.По мере взросления им может потребоваться трость или инвалидное кресло для передвижения.
Миотоническая дистрофия
Симптомы миотонической дистрофии могут быть очевидны с рождения или развиваться позже — в подростковом или взрослом возрасте.
Как и другие формы мышечной дистрофии, миотоническая дистрофия приводит к мышечной слабости, которая со временем ухудшается, что приводит к неспособности намеренно расслабить мышцы. Обычно сначала поражаются мелкие мышцы, например:
Симптомы миотонической дистрофии могут появиться в любой момент жизни человека.Симптомы включают:
- Слабость мышц лица, рук и шеи
- Мышечная ригидность (миотония) — трудности с расслаблением мышц после их напряжения
- Сокращение мышц с течением времени (мышечное истощение)
- Катаракта — помутнение хрусталика глаза
- Дневная сонливость
- Проблемы с обучением и поведением
- Проблемы с сердцем, включая нерегулярное сердцебиение (аритмию)
Тип миотонической дистрофии, которая начинается при рождении, более тяжелый.Другие формы ухудшаются очень медленно, и на прогрессирование может уйти от 50 до 60 лет.
Конечностно-поясная мышечная дистрофия
Эта форма мышечной дистрофии фактически представляет собой группу связанных состояний. Обычно это начинается в детстве или в подростковом возрасте.
Часто мышцы, которые сначала слабеют, оказываются большими мышцами:
Слабость мышц со временем ухудшается очень медленно.
Другие симптомы включают:
- Потеря мышц в пораженных участках
- Боль в спине
- Проблемы с поднятием предметов
- Проблемы с бегом
- Учащенное сердцебиение (учащенное сердцебиение) или нерегулярное сердцебиение
Насколько серьезны последствия, зависит от ребенка .У некоторых детей наблюдается лишь небольшая мышечная слабость. Другие настолько слабы, что им необходимо пользоваться инвалидной коляской.
На более поздних стадиях мышечная дистрофия конечностей и поясов может вызвать серьезные проблемы с сердцем.