причины, симптомы, диагностика и лечение
Глухонемота (сурдомутизм) – врожденное или приобретенное отсутствие слуха, произошедшее до момента, когда ребенок начнет говорить. Проявляется немотой, глухотой, искажением голоса, нарушениями поведения, сложностями в удерживании равновесия, очень развитой мимикой. Приобретенная форма болезни встречается более чем в половине случаев. Чаще встречается у детей, рожденных от близкородственных браков.
Причины
Первым всегда теряется слух, а отсутствие речи – это косвенный признак, возникающий из-за невозможности изучать слова, звуки и воспроизводить их. Причины врожденной глухонемоты:
- TORCH-инфекции во время беременности (токсоплазмоз, краснуха, цитомегаловирус, герпес-инфекция, сифилис, гепатит В).
- Генные мутации (синдром Ваарденбурга, Стиклера, Пендреда, Альпорта, нейрофиброматоз, дисплазия Мондини, болезнь Рефсума и другие).
- Интоксикация плода (алкоголизм матери, наркотическая зависимость, работа с химическими агентами).

Причины приобретенной глухонемоты:
- Родовая травма.
- Поражение слухового нерва или структур внутреннего уха (лабиринтит, болезнь Меньера, кохлеарный неврит и др.).
- Прием ототоксичных лекарств (фторхинолоны, петлевые диуретики, аминогликозиды).
- Инфекции (менингит, скарлатина, корь, брюшной тип, дифтерия, коклюш, сифилис).
Симптомы
Если не было предрасполагающих факторов, которые подтолкнут родителей к определению слуха сразу после рождения ребенка, то глухота долгое время может оставаться незамеченной. В первые месяцы жизни дети плачут и кричат рефлекторно и только к полугодовалому возрасту появляются попытки имитации отдельных звуков.
При глухонемоте данный порядок развития нарушается, взамен отсутствующей речи, активно развивается мимика ребенка. Он шевелит губами, пытаясь скопировать движения родителей. Если забоелвание возникло позже 2 лет, то человек перестает реагировать на свое имя, посторонние звуки, музыку. Дети старшего возраста могут жаловаться на шум в ушах или внезапное снижение слуха. Речь, которая успела сформироваться до этого периода, искажается: становится тихая или слишком громкая, монотонная, меняется тембр голоса. Со временем прогрессируют психические нарушения аутического спектра (отчужденность, замкнутость, раздражительность, вспыльчивость).
Речь, которая успела сформироваться до этого периода, искажается: становится тихая или слишком громкая, монотонная, меняется тембр голоса. Со временем прогрессируют психические нарушения аутического спектра (отчужденность, замкнутость, раздражительность, вспыльчивость).
Долгое отсутствие речевой активность приводит к морфологическим изменениям гортани:
— голосовые связки смыкаются не полностью;
— рано окостеневают хрящи.
Без полноценной и своевременной реабилитации дети не могут адаптироваться к жизни в социуме.
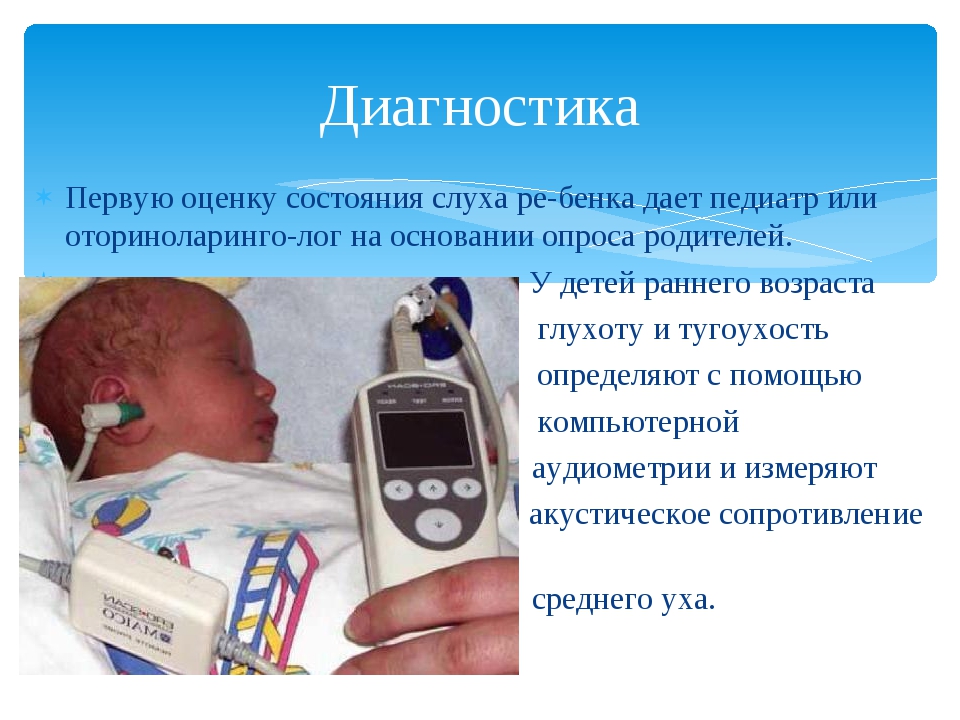
Диагностика
Сурдологи и оториноларингологи проверяют функционирование звукопроводящего и звуковоспринимающего аппаратов. Для этого они используют следующие тесты:
- Исследование звукового восприятия (отклик на разные по тембру и громкости звуки).
- ABR-тест (функция слуховых нервов и ядер ствола мозга).
- Нейровизуализация (КТ, МРТ).
- Генетическая диагностика.
Лечение
Как правило, восстановить слух до нормального уровня при сформировавшейся глухонемоте уже не получится. Если заболевание приобретенное, то на ранних его этапах еще можно провести консервативное или оперативное лечение. При врожденном сурдомутизме детям рекомендуется заниматься по специальной развивающей программе. Педагоги обучают ребенка языку жестов, чтению по губам и другим техникам социализации. Помимо этого, возможно применение слуховых аппаратов и имплантов внутреннего уха.
Если заболевание приобретенное, то на ранних его этапах еще можно провести консервативное или оперативное лечение. При врожденном сурдомутизме детям рекомендуется заниматься по специальной развивающей программе. Педагоги обучают ребенка языку жестов, чтению по губам и другим техникам социализации. Помимо этого, возможно применение слуховых аппаратов и имплантов внутреннего уха.
Прогноз течения заболевания неблагоприятный, потеря слуха необратима. Как профилактические меры можно использовать медико-генетическое консультирование, тщательное планирование беременности, ранняя диагностика и лечение инфекционных заболеваний.
причины, симптомы, диагностика и лечение
Глухонемота, или сурдомутизм – это врожденное или приобретенное в долингвальном периоде отсутствие слуха, которое препятствует развитию речи. Основные клинические проявления – глухота, немота, искажение голоса, вестибулярные и поведенческие нарушения, чрезмерно развитая мимика. Постановка диагноза основывается на данных исследования звукового восприятия, ABR-тесте, методах нейровизуализации и ДНК-диагностике. Специфическое лечение отсутствует. Реабилитационные мероприятия включают в себя слухопротезирование, изучение языка жестов и артикуляции.
Постановка диагноза основывается на данных исследования звукового восприятия, ABR-тесте, методах нейровизуализации и ДНК-диагностике. Специфическое лечение отсутствует. Реабилитационные мероприятия включают в себя слухопротезирование, изучение языка жестов и артикуляции.
Общие сведения
Существует две основные формы глухонемоты – врожденная и приобретенная. Второй вариант встречается более чем у 70% от общего количества больных. Свыше 50% случаев врожденной формы заболевания спровоцировано генетическими мутациями. Согласно статистическим данным мировой отоларингологии, распространенность глухонемоты выше на территориях с высокой частотой близкородственных браков. Порядка 35% случаев приобретенной потери слуха происходит на первом году жизни ребенка, 25% – на втором. Мальчики и девочки болеют с одинаковой частотой. Всего на территории Российской Федерации проживает порядка 200 тысяч глухонемых.
Глухонемота
Причины глухонемоты
Первопричиной всегда является врожденная или приобретенная в раннем возрасте потеря слуха. Немота возникает вторично в результате неспособности изучать и воспроизводить слова. Врожденная глухонемота может быть спровоцирована следующими факторами:
Немота возникает вторично в результате неспособности изучать и воспроизводить слова. Врожденная глухонемота может быть спровоцирована следующими факторами:
- Врожденные инфекции. Чаще всего это заболевания из группы TORCH-инфекций: токсоплазмоз, краснуха, цитомегалия, герпетическая инфекция, сифилис, гепатит В.
- Генетические мутации. Глухота входит в состав более 400 генетических синдромов. Наиболее распространенными являются синдромы Ваарденбурга, Стиклера, Пендреда, Альпорта, брахио-ото-ренальный синдром, нейрофиброматоз II типа, дисплазия Мондини, болезнь Рефсума и недостаточность биотинидазы.
- Внутриутробная интоксикация. Врожденное отсутствие слуха может вызывать злоупотребление алкоголем и наркотическими веществами во время вынашивания ребенка, а также контакт с химическими веществами в условиях производства.
В случаях приобретенной глухонемоты слух у ребенка на момент рождения присутствует, но через короткий промежуток времени резко ухудшается или полностью исчезает.
- Родовая травма новорожденного. Неправильно выбранный метод родоразрешения и некорректное использование акушерских пособий может привести к повреждению анатомических структур среднего и внутреннего уха, корковых центров головного мозга.
- Поражение внутреннего уха, слухового нерва. Рецидивирующий лабиринтит, осложненное течение болезни Меньера, двухсторонний кохлеарный неврит и врожденные аномалии развития внутреннего уха могут становиться причиной нарушения функции кортиевого органа и, как следствие – глухоты.
- Ототоксические медикаменты. Потеря слуха может выступать в роли побочного эффекта при приеме антибиотиков из групп полипептидов и полимиксинов, некоторых аминогликозидов и петлевых диуретиков.
- Инфекционные заболевания. К глухоте может приводить перенесенный в детском возрасте цереброспинальный менингит, скарлатина, корь, брюшной тиф, сифилис, грипп, дифтерия и коклюш.

Патогенез
Ведущее нарушение при глухонемоте – стойкое ухудшение слуха или его полное отсутствие. Причинами глухоты могут быть структурные аномалии, инфекционные и воспалительные заболевания внутреннего уха или непосредственно звуковоспринимающего аппарата (кортиевого органа), слухового нерва и задних отделов верхней височной извилины – области Вернике. Речевые центры (лобная извилина – зона Брока) и органы артикуляционного аппарата не поражены. При врожденной глухоте ребенок с рождения не воспринимает окружающие звуки, в том числе человеческую речь, что делает невозможным ее изучение – формируется вторичная немота. При потере слуха в возрасте старше 1 года дети уже имеют некоторый словарный запас, но при отсутствии специальных мероприятий по развитию речи приобретенные навыки быстро теряются.
Симптомы глухонемоты
При отсутствии прицельной диагностики слуха у новорожденного заболевание долгое время может оставаться незамеченным. На первых месяцах жизни у детей проявляется рефлекторный плач и крик за счет нормально функционирующих ЦНС и органов речевого аппарата. В норме в возрасте 6-7 месяцев ребенок начинает имитировать речь окружающих и выговаривать первые слоги («ма-ма», «па-па»), а в 1 год его словарный запас составляет порядка полноценных 10 слов. При глухонемоте подобная речевая активность полностью отсутствует, компенсаторно сильно развивается мимика. В некоторых случаях ребенок издает отдельные звуки в попытке имитировать движение губ родителей.
На первых месяцах жизни у детей проявляется рефлекторный плач и крик за счет нормально функционирующих ЦНС и органов речевого аппарата. В норме в возрасте 6-7 месяцев ребенок начинает имитировать речь окружающих и выговаривать первые слоги («ма-ма», «па-па»), а в 1 год его словарный запас составляет порядка полноценных 10 слов. При глухонемоте подобная речевая активность полностью отсутствует, компенсаторно сильно развивается мимика. В некоторых случаях ребенок издает отдельные звуки в попытке имитировать движение губ родителей.
При приобретенной глухонемоте, возникшей у детей старше 2 лет, ребенок резко прекращает воспринимать внешние звуки – не откликается на свое имя, не реагирует на музыку и т. д. В 3-4 года дети могут предъявлять жалобы на шум в ушах или резкую потерю слуха. Одновременно искажается уже сформированная речь – она становится чрезмерно громкой или тихой, скандированной, монотонной. У некоторых детей формируется нетипичный для пола и возраста низкий или высокий тембр голоса. Выраженность вестибулярных расстройств напрямую зависит от этиопатогенетического варианта глухонемоты. Зачастую они ограничиваются плохим чувством равновесия, особенно в условиях темноты или с закрытыми глазами. В возрасте после 3 лет возникают психические нарушения – замкнутость, отчуждение, вспыльчивость и раздражительность. В редких случаях наблюдаются противоположные изменения поведения – чрезмерная веселость, коммуникабельность и подвижность.
Выраженность вестибулярных расстройств напрямую зависит от этиопатогенетического варианта глухонемоты. Зачастую они ограничиваются плохим чувством равновесия, особенно в условиях темноты или с закрытыми глазами. В возрасте после 3 лет возникают психические нарушения – замкнутость, отчуждение, вспыльчивость и раздражительность. В редких случаях наблюдаются противоположные изменения поведения – чрезмерная веселость, коммуникабельность и подвижность.
Осложнения
Отсутствие речевой активности при глухонемоте сопровождается неправильным функционированием голосового аппарата. В дальнейшем это приводит морфологическим изменениям гортани – неполному смыканию голосовой щели, преждевременному окостенению хрящей и т. д. Это делает невозможным формирование естественного звучания речи даже на фоне ее поддержания и дальнейшего развития. Отсутствие полноценных реабилитационных мер и обучения в специализированных учреждениях делает практически невозможной полноценную адаптацию пациента в социуме.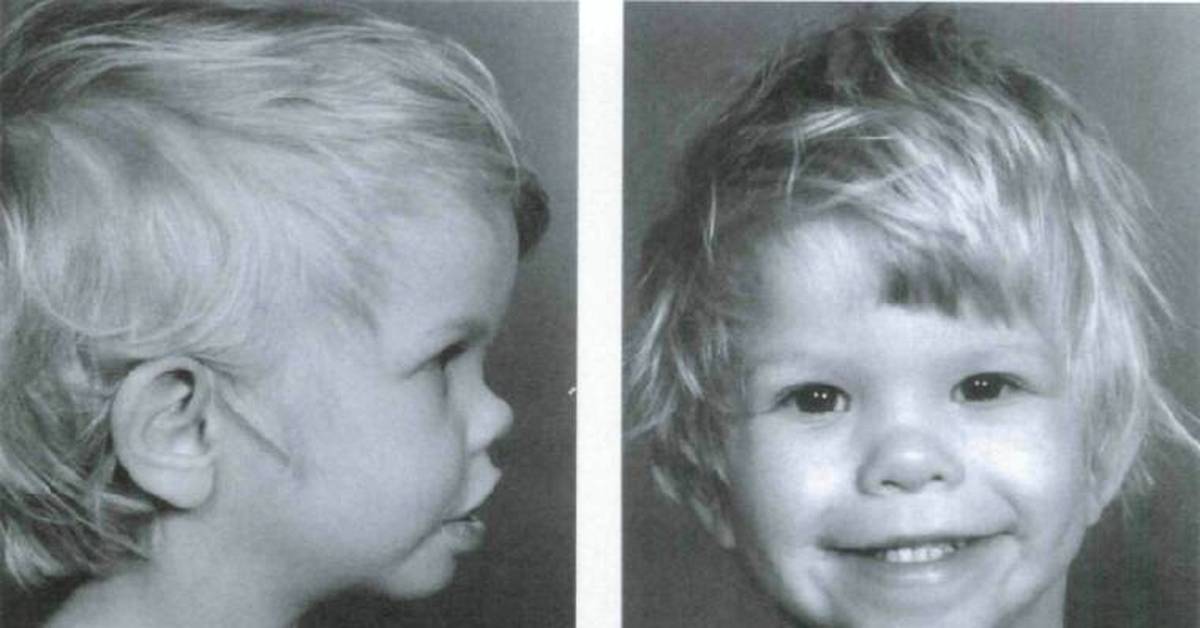
Диагностика
Диагностика при глухонемоте заключается в исследовании звукопроводящего и звуковоспринимающего аппаратов, изучении структуры височных и лобных отделов коры головного мозга и установлении этиологического фактора. Сложность обследования детей до 2 лет состоит в том, что распространенные тесты с использованием камертона и аудиометра непонятны для ребенка и не дают возможности получить четкие результаты. Таким образом, диагностическая программа включает:
- Исследование звукового восприятия. Отоларинголог при осмотре пациента разговаривает с ним с разной громкостью, дает ему звучащие игрушки. Для исключения тактильных ощущений от выдыхаемого воздуха или визуального восприятия движения губ врач использует маску или лист бумаги. При глухоте у ребенка отсутствует какая-либо реакция на окружающие звуки.
- ABR-тест. Позволяет оценить функцию проводящих нервов и слухового отдела ствола мозга. При их поражении нервный импульс из внутреннего уха не передается или не воспринимается структурами ЦНС.
- Нейровизуализацию. КТ черепа, МРТ височных костей и МРТ головного мозга позволяют выявить структурные или воспалительные изменения в строении звукопроводящей и звуковоспринимающей систем, мозговой коры.
- ДНК-диагностику. Изучение структуры ДНК применяется при наличии других симптомов, указывающих на развитие того или иного генетического заболевания.
 При их поражении нервный импульс из внутреннего уха не передается или не воспринимается структурами ЦНС.
При их поражении нервный импульс из внутреннего уха не передается или не воспринимается структурами ЦНС.Лечение глухонемоты
В большинстве случаев восстановить нормальный уровень слуха при условии уже сформировавшегося сурдомутизма невозможно. На ранних этапах прогрессирующего ухудшения звуковосприятия проводится медикаментозное или хирургическое лечение с учетом этиологических факторов. Дети с врожденной глухотой направляются в специализированные воспитательные и учебные заведения. При глухонемоте, сформировавшейся в возрасте 3-5 лет, осуществляется дальнейшее развитие речи при помощи занятий с сурдопедагогом. К реабилитационным мероприятиям относится применение слуховых аппаратов и кохлеарных имплантатов. Эти приборы эффективны при глухоте, возникшей в результате поражения органов звукопроводящей системы или внутреннего уха.
К реабилитационным мероприятиям относится применение слуховых аппаратов и кохлеарных имплантатов. Эти приборы эффективны при глухоте, возникшей в результате поражения органов звукопроводящей системы или внутреннего уха.
Прогноз и профилактика
Прогноз при глухонемоте неблагоприятный. В подавляющем большинстве случаев потеря слуха – это проявление финальной стадии заболевания, на которой изменения уже необратимы. В дальнейшем дети проходят специальное обучение, направленное на развитие частично сформированной речи или изучение артикуляции и языка жестов. К профилактическим мероприятиям относится медико-генетическое консультирование семейных пар, планирование беременности, антенатальная охрана плода, ранняя диагностика и лечение заболеваний, которые потенциально могут привести к глухоте.
причины, симптомы, диагностика и лечение
Глухонемота, или сурдомутизм – это врожденное или приобретенное в долингвальном периоде отсутствие слуха, которое препятствует развитию речи. Основные клинические проявления – глухота, немота, искажение голоса, вестибулярные и поведенческие нарушения, чрезмерно развитая мимика. Постановка диагноза основывается на данных исследования звукового восприятия, ABR-тесте, методах нейровизуализации и ДНК-диагностике. Специфическое лечение отсутствует. Реабилитационные мероприятия включают в себя слухопротезирование, изучение языка жестов и артикуляции.
Основные клинические проявления – глухота, немота, искажение голоса, вестибулярные и поведенческие нарушения, чрезмерно развитая мимика. Постановка диагноза основывается на данных исследования звукового восприятия, ABR-тесте, методах нейровизуализации и ДНК-диагностике. Специфическое лечение отсутствует. Реабилитационные мероприятия включают в себя слухопротезирование, изучение языка жестов и артикуляции.
Общие сведения
Существует две основные формы глухонемоты – врожденная и приобретенная. Второй вариант встречается более чем у 70% от общего количества больных. Свыше 50% случаев врожденной формы заболевания спровоцировано генетическими мутациями. Согласно статистическим данным мировой отоларингологии, распространенность глухонемоты выше на территориях с высокой частотой близкородственных браков. Порядка 35% случаев приобретенной потери слуха происходит на первом году жизни ребенка, 25% – на втором. Мальчики и девочки болеют с одинаковой частотой. Всего на территории Российской Федерации проживает порядка 200 тысяч глухонемых.
Всего на территории Российской Федерации проживает порядка 200 тысяч глухонемых.
Глухонемота
Причины глухонемоты
Первопричиной всегда является врожденная или приобретенная в раннем возрасте потеря слуха. Немота возникает вторично в результате неспособности изучать и воспроизводить слова. Врожденная глухонемота может быть спровоцирована следующими факторами:
- Врожденные инфекции. Чаще всего это заболевания из группы TORCH-инфекций: токсоплазмоз, краснуха, цитомегалия, герпетическая инфекция, сифилис, гепатит В.
- Генетические мутации. Глухота входит в состав более 400 генетических синдромов. Наиболее распространенными являются синдромы Ваарденбурга, Стиклера, Пендреда, Альпорта, брахио-ото-ренальный синдром, нейрофиброматоз II типа, дисплазия Мондини, болезнь Рефсума и недостаточность биотинидазы.
- Внутриутробная интоксикация. Врожденное отсутствие слуха может вызывать злоупотребление алкоголем и наркотическими веществами во время вынашивания ребенка, а также контакт с химическими веществами в условиях производства.

В случаях приобретенной глухонемоты слух у ребенка на момент рождения присутствует, но через короткий промежуток времени резко ухудшается или полностью исчезает. Причиной может быть:
- Родовая травма новорожденного. Неправильно выбранный метод родоразрешения и некорректное использование акушерских пособий может привести к повреждению анатомических структур среднего и внутреннего уха, корковых центров головного мозга.
- Поражение внутреннего уха, слухового нерва. Рецидивирующий лабиринтит, осложненное течение болезни Меньера, двухсторонний кохлеарный неврит и врожденные аномалии развития внутреннего уха могут становиться причиной нарушения функции кортиевого органа и, как следствие – глухоты.
- Ототоксические медикаменты. Потеря слуха может выступать в роли побочного эффекта при приеме антибиотиков из групп полипептидов и полимиксинов, некоторых аминогликозидов и петлевых диуретиков.
- Инфекционные заболевания. К глухоте может приводить перенесенный в детском возрасте цереброспинальный менингит, скарлатина, корь, брюшной тиф, сифилис, грипп, дифтерия и коклюш.

Патогенез
Ведущее нарушение при глухонемоте – стойкое ухудшение слуха или его полное отсутствие. Причинами глухоты могут быть структурные аномалии, инфекционные и воспалительные заболевания внутреннего уха или непосредственно звуковоспринимающего аппарата (кортиевого органа), слухового нерва и задних отделов верхней височной извилины – области Вернике. Речевые центры (лобная извилина – зона Брока) и органы артикуляционного аппарата не поражены. При врожденной глухоте ребенок с рождения не воспринимает окружающие звуки, в том числе человеческую речь, что делает невозможным ее изучение – формируется вторичная немота. При потере слуха в возрасте старше 1 года дети уже имеют некоторый словарный запас, но при отсутствии специальных мероприятий по развитию речи приобретенные навыки быстро теряются.
Симптомы глухонемоты
При отсутствии прицельной диагностики слуха у новорожденного заболевание долгое время может оставаться незамеченным. На первых месяцах жизни у детей проявляется рефлекторный плач и крик за счет нормально функционирующих ЦНС и органов речевого аппарата. В норме в возрасте 6-7 месяцев ребенок начинает имитировать речь окружающих и выговаривать первые слоги («ма-ма», «па-па»), а в 1 год его словарный запас составляет порядка полноценных 10 слов. При глухонемоте подобная речевая активность полностью отсутствует, компенсаторно сильно развивается мимика. В некоторых случаях ребенок издает отдельные звуки в попытке имитировать движение губ родителей.
При приобретенной глухонемоте, возникшей у детей старше 2 лет, ребенок резко прекращает воспринимать внешние звуки – не откликается на свое имя, не реагирует на музыку и т. д. В 3-4 года дети могут предъявлять жалобы на шум в ушах или резкую потерю слуха. Одновременно искажается уже сформированная речь – она становится чрезмерно громкой или тихой, скандированной, монотонной. У некоторых детей формируется нетипичный для пола и возраста низкий или высокий тембр голоса. Выраженность вестибулярных расстройств напрямую зависит от этиопатогенетического варианта глухонемоты. Зачастую они ограничиваются плохим чувством равновесия, особенно в условиях темноты или с закрытыми глазами. В возрасте после 3 лет возникают психические нарушения – замкнутость, отчуждение, вспыльчивость и раздражительность. В редких случаях наблюдаются противоположные изменения поведения – чрезмерная веселость, коммуникабельность и подвижность.
У некоторых детей формируется нетипичный для пола и возраста низкий или высокий тембр голоса. Выраженность вестибулярных расстройств напрямую зависит от этиопатогенетического варианта глухонемоты. Зачастую они ограничиваются плохим чувством равновесия, особенно в условиях темноты или с закрытыми глазами. В возрасте после 3 лет возникают психические нарушения – замкнутость, отчуждение, вспыльчивость и раздражительность. В редких случаях наблюдаются противоположные изменения поведения – чрезмерная веселость, коммуникабельность и подвижность.
Осложнения
Отсутствие речевой активности при глухонемоте сопровождается неправильным функционированием голосового аппарата. В дальнейшем это приводит морфологическим изменениям гортани – неполному смыканию голосовой щели, преждевременному окостенению хрящей и т. д. Это делает невозможным формирование естественного звучания речи даже на фоне ее поддержания и дальнейшего развития. Отсутствие полноценных реабилитационных мер и обучения в специализированных учреждениях делает практически невозможной полноценную адаптацию пациента в социуме.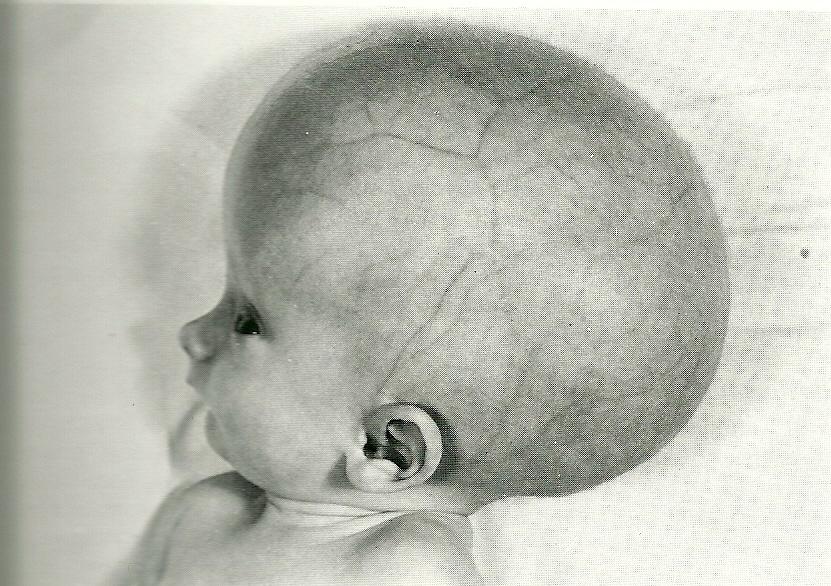
Диагностика
Диагностика при глухонемоте заключается в исследовании звукопроводящего и звуковоспринимающего аппаратов, изучении структуры височных и лобных отделов коры головного мозга и установлении этиологического фактора. Сложность обследования детей до 2 лет состоит в том, что распространенные тесты с использованием камертона и аудиометра непонятны для ребенка и не дают возможности получить четкие результаты. Таким образом, диагностическая программа включает:
- Исследование звукового восприятия. Отоларинголог при осмотре пациента разговаривает с ним с разной громкостью, дает ему звучащие игрушки. Для исключения тактильных ощущений от выдыхаемого воздуха или визуального восприятия движения губ врач использует маску или лист бумаги. При глухоте у ребенка отсутствует какая-либо реакция на окружающие звуки.
- ABR-тест. Позволяет оценить функцию проводящих нервов и слухового отдела ствола мозга. При их поражении нервный импульс из внутреннего уха не передается или не воспринимается структурами ЦНС.
- Нейровизуализацию. КТ черепа, МРТ височных костей и МРТ головного мозга позволяют выявить структурные или воспалительные изменения в строении звукопроводящей и звуковоспринимающей систем, мозговой коры.
- ДНК-диагностику. Изучение структуры ДНК применяется при наличии других симптомов, указывающих на развитие того или иного генетического заболевания.
 При их поражении нервный импульс из внутреннего уха не передается или не воспринимается структурами ЦНС.
При их поражении нервный импульс из внутреннего уха не передается или не воспринимается структурами ЦНС.Лечение глухонемоты
В большинстве случаев восстановить нормальный уровень слуха при условии уже сформировавшегося сурдомутизма невозможно. На ранних этапах прогрессирующего ухудшения звуковосприятия проводится медикаментозное или хирургическое лечение с учетом этиологических факторов. Дети с врожденной глухотой направляются в специализированные воспитательные и учебные заведения. При глухонемоте, сформировавшейся в возрасте 3-5 лет, осуществляется дальнейшее развитие речи при помощи занятий с сурдопедагогом. К реабилитационным мероприятиям относится применение слуховых аппаратов и кохлеарных имплантатов. Эти приборы эффективны при глухоте, возникшей в результате поражения органов звукопроводящей системы или внутреннего уха.
К реабилитационным мероприятиям относится применение слуховых аппаратов и кохлеарных имплантатов. Эти приборы эффективны при глухоте, возникшей в результате поражения органов звукопроводящей системы или внутреннего уха.
Прогноз и профилактика
Прогноз при глухонемоте неблагоприятный. В подавляющем большинстве случаев потеря слуха – это проявление финальной стадии заболевания, на которой изменения уже необратимы. В дальнейшем дети проходят специальное обучение, направленное на развитие частично сформированной речи или изучение артикуляции и языка жестов. К профилактическим мероприятиям относится медико-генетическое консультирование семейных пар, планирование беременности, антенатальная охрана плода, ранняя диагностика и лечение заболеваний, которые потенциально могут привести к глухоте.
причины, симптомы, диагностика и лечение
Глухонемота, или сурдомутизм – это врожденное или приобретенное в долингвальном периоде отсутствие слуха, которое препятствует развитию речи. Основные клинические проявления – глухота, немота, искажение голоса, вестибулярные и поведенческие нарушения, чрезмерно развитая мимика. Постановка диагноза основывается на данных исследования звукового восприятия, ABR-тесте, методах нейровизуализации и ДНК-диагностике. Специфическое лечение отсутствует. Реабилитационные мероприятия включают в себя слухопротезирование, изучение языка жестов и артикуляции.
Основные клинические проявления – глухота, немота, искажение голоса, вестибулярные и поведенческие нарушения, чрезмерно развитая мимика. Постановка диагноза основывается на данных исследования звукового восприятия, ABR-тесте, методах нейровизуализации и ДНК-диагностике. Специфическое лечение отсутствует. Реабилитационные мероприятия включают в себя слухопротезирование, изучение языка жестов и артикуляции.
Общие сведения
Существует две основные формы глухонемоты – врожденная и приобретенная. Второй вариант встречается более чем у 70% от общего количества больных. Свыше 50% случаев врожденной формы заболевания спровоцировано генетическими мутациями. Согласно статистическим данным мировой отоларингологии, распространенность глухонемоты выше на территориях с высокой частотой близкородственных браков. Порядка 35% случаев приобретенной потери слуха происходит на первом году жизни ребенка, 25% – на втором. Мальчики и девочки болеют с одинаковой частотой. Всего на территории Российской Федерации проживает порядка 200 тысяч глухонемых.
Всего на территории Российской Федерации проживает порядка 200 тысяч глухонемых.
Глухонемота
Причины глухонемоты
Первопричиной всегда является врожденная или приобретенная в раннем возрасте потеря слуха. Немота возникает вторично в результате неспособности изучать и воспроизводить слова. Врожденная глухонемота может быть спровоцирована следующими факторами:
- Врожденные инфекции. Чаще всего это заболевания из группы TORCH-инфекций: токсоплазмоз, краснуха, цитомегалия, герпетическая инфекция, сифилис, гепатит В.
- Генетические мутации. Глухота входит в состав более 400 генетических синдромов. Наиболее распространенными являются синдромы Ваарденбурга, Стиклера, Пендреда, Альпорта, брахио-ото-ренальный синдром, нейрофиброматоз II типа, дисплазия Мондини, болезнь Рефсума и недостаточность биотинидазы.
- Внутриутробная интоксикация. Врожденное отсутствие слуха может вызывать злоупотребление алкоголем и наркотическими веществами во время вынашивания ребенка, а также контакт с химическими веществами в условиях производства.

В случаях приобретенной глухонемоты слух у ребенка на момент рождения присутствует, но через короткий промежуток времени резко ухудшается или полностью исчезает. Причиной может быть:
- Родовая травма новорожденного. Неправильно выбранный метод родоразрешения и некорректное использование акушерских пособий может привести к повреждению анатомических структур среднего и внутреннего уха, корковых центров головного мозга.
- Поражение внутреннего уха, слухового нерва. Рецидивирующий лабиринтит, осложненное течение болезни Меньера, двухсторонний кохлеарный неврит и врожденные аномалии развития внутреннего уха могут становиться причиной нарушения функции кортиевого органа и, как следствие – глухоты.
- Ототоксические медикаменты. Потеря слуха может выступать в роли побочного эффекта при приеме антибиотиков из групп полипептидов и полимиксинов, некоторых аминогликозидов и петлевых диуретиков.
- Инфекционные заболевания. К глухоте может приводить перенесенный в детском возрасте цереброспинальный менингит, скарлатина, корь, брюшной тиф, сифилис, грипп, дифтерия и коклюш.
Патогенез
Ведущее нарушение при глухонемоте – стойкое ухудшение слуха или его полное отсутствие. Причинами глухоты могут быть структурные аномалии, инфекционные и воспалительные заболевания внутреннего уха или непосредственно звуковоспринимающего аппарата (кортиевого органа), слухового нерва и задних отделов верхней височной извилины – области Вернике. Речевые центры (лобная извилина – зона Брока) и органы артикуляционного аппарата не поражены. При врожденной глухоте ребенок с рождения не воспринимает окружающие звуки, в том числе человеческую речь, что делает невозможным ее изучение – формируется вторичная немота. При потере слуха в возрасте старше 1 года дети уже имеют некоторый словарный запас, но при отсутствии специальных мероприятий по развитию речи приобретенные навыки быстро теряются.
Симптомы глухонемоты
При отсутствии прицельной диагностики слуха у новорожденного заболевание долгое время может оставаться незамеченным. На первых месяцах жизни у детей проявляется рефлекторный плач и крик за счет нормально функционирующих ЦНС и органов речевого аппарата. В норме в возрасте 6-7 месяцев ребенок начинает имитировать речь окружающих и выговаривать первые слоги («ма-ма», «па-па»), а в 1 год его словарный запас составляет порядка полноценных 10 слов. При глухонемоте подобная речевая активность полностью отсутствует, компенсаторно сильно развивается мимика. В некоторых случаях ребенок издает отдельные звуки в попытке имитировать движение губ родителей.
При приобретенной глухонемоте, возникшей у детей старше 2 лет, ребенок резко прекращает воспринимать внешние звуки – не откликается на свое имя, не реагирует на музыку и т. д. В 3-4 года дети могут предъявлять жалобы на шум в ушах или резкую потерю слуха. Одновременно искажается уже сформированная речь – она становится чрезмерно громкой или тихой, скандированной, монотонной. У некоторых детей формируется нетипичный для пола и возраста низкий или высокий тембр голоса. Выраженность вестибулярных расстройств напрямую зависит от этиопатогенетического варианта глухонемоты. Зачастую они ограничиваются плохим чувством равновесия, особенно в условиях темноты или с закрытыми глазами. В возрасте после 3 лет возникают психические нарушения – замкнутость, отчуждение, вспыльчивость и раздражительность. В редких случаях наблюдаются противоположные изменения поведения – чрезмерная веселость, коммуникабельность и подвижность.
Осложнения
Отсутствие речевой активности при глухонемоте сопровождается неправильным функционированием голосового аппарата. В дальнейшем это приводит морфологическим изменениям гортани – неполному смыканию голосовой щели, преждевременному окостенению хрящей и т. д. Это делает невозможным формирование естественного звучания речи даже на фоне ее поддержания и дальнейшего развития. Отсутствие полноценных реабилитационных мер и обучения в специализированных учреждениях делает практически невозможной полноценную адаптацию пациента в социуме.
Диагностика
Диагностика при глухонемоте заключается в исследовании звукопроводящего и звуковоспринимающего аппаратов, изучении структуры височных и лобных отделов коры головного мозга и установлении этиологического фактора. Сложность обследования детей до 2 лет состоит в том, что распространенные тесты с использованием камертона и аудиометра непонятны для ребенка и не дают возможности получить четкие результаты. Таким образом, диагностическая программа включает:
- Исследование звукового восприятия. Отоларинголог при осмотре пациента разговаривает с ним с разной громкостью, дает ему звучащие игрушки. Для исключения тактильных ощущений от выдыхаемого воздуха или визуального восприятия движения губ врач использует маску или лист бумаги. При глухоте у ребенка отсутствует какая-либо реакция на окружающие звуки.
- ABR-тест. Позволяет оценить функцию проводящих нервов и слухового отдела ствола мозга. При их поражении нервный импульс из внутреннего уха не передается или не воспринимается структурами ЦНС.
- Нейровизуализацию. КТ черепа, МРТ височных костей и МРТ головного мозга позволяют выявить структурные или воспалительные изменения в строении звукопроводящей и звуковоспринимающей систем, мозговой коры.
- ДНК-диагностику. Изучение структуры ДНК применяется при наличии других симптомов, указывающих на развитие того или иного генетического заболевания.
Лечение глухонемоты
В большинстве случаев восстановить нормальный уровень слуха при условии уже сформировавшегося сурдомутизма невозможно. На ранних этапах прогрессирующего ухудшения звуковосприятия проводится медикаментозное или хирургическое лечение с учетом этиологических факторов. Дети с врожденной глухотой направляются в специализированные воспитательные и учебные заведения. При глухонемоте, сформировавшейся в возрасте 3-5 лет, осуществляется дальнейшее развитие речи при помощи занятий с сурдопедагогом. К реабилитационным мероприятиям относится применение слуховых аппаратов и кохлеарных имплантатов. Эти приборы эффективны при глухоте, возникшей в результате поражения органов звукопроводящей системы или внутреннего уха.
Прогноз и профилактика
Прогноз при глухонемоте неблагоприятный. В подавляющем большинстве случаев потеря слуха – это проявление финальной стадии заболевания, на которой изменения уже необратимы. В дальнейшем дети проходят специальное обучение, направленное на развитие частично сформированной речи или изучение артикуляции и языка жестов. К профилактическим мероприятиям относится медико-генетическое консультирование семейных пар, планирование беременности, антенатальная охрана плода, ранняя диагностика и лечение заболеваний, которые потенциально могут привести к глухоте.
причины, симптомы, диагностика, лечение, профилактика
Второе название заболевания — сурдомутизм, это врожденное или приобретенное в долингвальном периоде отсутствие слуха, которое сопровождается нарушением развития речи.
ПричиныЗаболевание возникает на фоне врожденной или приобретенной в раннем возрасте глухоты. Практически во всех случаях развитие немоты является вторичным в результате неспособности изучать и воспроизводить слова. Врожденная глухонемота может возникать по самым разным причинам.
Довольно часто развитие данной патологии связывают с перенесенными женщиной инфекционными заболеваниями на стадии гестации. Чаще всего патология возникает в результате заболеваний из группы TORCH-инфекций, таких как токсоплазмоз, краснуха, цитомегалия, герпетическая инфекция, сифилис, гепатит В.
Глухота входит в состав более чем 400 генетических синдромов. Наиболее распространенными генетическими заболеваниями, на фоне которых может возникнуть глухота, считаются синдромы Ваарденбурга, Пендреда, Стиклера, Альпорта, дисплазия Мондини, болезнь Рефсума и недостаточность биотинидазы.
Внутриутробная интоксикация, обусловленная злоупотреблением алкоголем и наркотическими веществами во время вынашивания ребенка, а также контакт с химическими веществами в условиях производства также могут вызвать развитие данной патологии.
При приобретенной глухонемоте слух у ребенка на момент рождения присутствует, однако в короткое время резко ухудшается либо полностью исчезает. Глухота у ребенка может возникать в результате:
родовых травм, обоснованных некорректно выбранной тактикой родоразрешения или неправильным использованием акушерских приспособлений, что может привести к повреждению анатомических структур среднего и внутреннего уха, корковых центров головного мозга;
поражения внутреннего уха, слухового нерва, вызванного рецидивирующим лабиринтитом, осложненным течением болезни Меньера, двухсторонним кохлеарным невритом и врожденными аномалиями развития внутреннего уха, становшими причиной нарушения функции кортиевого органа и сопровождавшимися возникновением глухоты;
приема ототоксических лекарственных средств, таких как антибиотики из группы полипептидов и полимиксинов, некоторые аминогликозиды и петлевые диуретики;
перенесенных в детском возрасте инфекционных заболеваний, таких как цереброспинальный менингит, скарлатина, корь, брюшной тиф, сифилис, грипп, дифтерия и коклюш.
СимптомыПри отсутствии прицельной диагностики слуха у новорожденного данная патология может длительное время оставаться незамеченной. В первые месяцы жизни у детей возникает рефлекторный плач и крик за счет нормально функционирующих центральной нервной системы и органов речевого аппарата. В норме в возрасте 6 или 7 месяцев ребенок начинает имитировать речь окружающих и выговаривать первые слоги, а в 1 год его словарный запас составляет порядка полноценных 10 слов. При глухонемоте такая речевая активность полностью отсутствует, но компенсаторно у ребенка отмечается выраженное развитие мимики. Иногда ребенок может издавать отдельные звуки в попытке имитировать движение губ взрослых.
При приобретенной глухонемоте, которая возникла у ребенка старше 2 лет, малыш резко прекращает воспринимать внешние звуки: не откликается на свое имя, не реагирует на музыку. В 3 — 4 года такие малыши иногда жалуются на появление шума в ушах или резкую потерю слуха. Вместе с появлением глухоты искажается уже сформированная речь, она может становиться чрезмерно громкой либо тихой, скандированной и монотонной.
ДиагностикаДиагностика глухонемоты заключается в исследовании звукопроводящего и звуковоспринимающего аппаратов, изучении структуры височных и лобных отделов коры головного мозга и установлении этиологического фактора. Сложность обследования детей младше 2 лет заключается в том, что классические тесты с использованием камертона и аудиометра для малыша непонятны и не позволяют получить четкие результаты. Диагностирование глухонемоты у ребенка включает исследование звукового восприятия, ABR-тест, что позволяет оценить функцию проводящих нервов и слухового отдела ствола мозга. Может потребоваться проведение компьютерной или магниторезонансной томографии мозга и височной кости, что даст возможность выявить воспалительные или структурные изменения строения звукопроводящей системы.
ЛечениеВ большинстве случаев восстановить нормальный уровень слуха при уже сформировавшемся сурдомутизме невозможно. На ранних этапах прогрессирующего ухудшения звуковосприятия проводится медикаментозное или хирургическое лечение с учетом этиологических факторов. Дети, страдающие врожденной глухотой, направляются в специализированные воспитательные и учебные заведения. При глухонемоте, сформировавшейся в возрасте 3 или 5 лет, осуществляется дальнейшее развитие речи при помощи занятий с сурдопедагогом. К реабилитационным мероприятиям относится применение слуховых аппаратов и кохлеарных имплантатов, которые эффективны при глухоте, возникшей в результате поражения органов звукопроводящей системы или внутреннего уха.
ПрофилактикаПрофилактика сурдомутизма основывается на проведении медико-генетических консультаций семейных пар с неблагоприятным семейным анамнезом.
С момента рождения или в раннем возрасте вследствие нарушения слуха, если ребенок перестал слышать, еще не овладев навыками речи, у него развивается глухонемота. Врожденная глухонемота может иметь наследственный характер, являться признаком отсутствия, недоразвития или разрушения слухового нерва. У детей с такой патологией гортань и прочие ЛОР-органы обычно не изменены. Приобретенная глухота вызывается заболеваниями уха, инфекционными болезнями, эпидемическим цереброспинальным менингитом, менингогенным лабиринтом, сифилисом, травмами, интоксикацией. Эти причины могут помешать ребенку, потерявшему слух, овладеть речью.
Чаще всего глухонемота бывает неполной. И при врожденной, и при приобретенной глухонемоте имеются остатки слуха. В барабанной перепонке каких-либо изменений обычно не наблюдается. Распознать глухонемоту у новорожденных и детишек раннего возраста обычно сложно, но возможно. Чаще всего она проявляется, когда ребенок начинает пытаться говорить. Если малыш реагирует на упавший предмет, это не всегда означает, что со слухом у него все в порядке, поскольку в данном случае ребенка привлекает не шум, а раздражение от сотрясения.
У ребенка, который до того, как оглохнуть, уже умел разговаривать, речь становится неясной, беззвучной, монотонной. Отсутствие слухового контроля ведет к развитию патологических изменений в артикуляционном аппарате, нарушению дыхательных и фонаторных функций.
Если в ухе произошли необратимые изменения, вылечить глухоту невозможно. Однако можно восстановить или улучшить речь. Многие глухонемые дети заканчивают обычные учебные заведения и становятся полноправными членами общества. Остатки слуха нужно использовать как можно раньше, иначе они полностью пропадают, что приводит к инвалидности. При наличии остатков слуха, лучшим помощником в данной ситуации будет слуховой аппарат. Из моего личного опыта, дети, имевшие серьезные нарушения речи и ходившие в специальную школу, которым одевали слуховые аппараты только в 11-13 лет, в кратчайшие сроки начинали говорить нормально и могли перейти в обычную школу. Конечно, при полной глухоте это невозможно, и шансы помочь уменьшаются в геометрической прогрессии, с затягиванием со слухопротезированием, но полная глухота встречается редко. Чаще встречается недостаток информации у родителей, отсутствие финансов на хороший слуховой аппарат, безразличие общества (так как помогать здесь надо финансово и в полном объеме, а не словами или полумерами), а иногда и самих родителей к данной проблеме. Есть также сложности и технического плана, когда из-за ограниченных возможностей берут низкобюджетные аппараты детям, аппарат вроде есть, но ребенок категорически отказывается его носить из-за сложности адаптации к такому звуку. Такое случается, когда носят и премиум аппараты, но там исправить ситуацию значительно легче, если провести все необходимые тесты для настройки слухового аппарата, которые можно сделать, когда ребенок станет старше.
Упражнения по восстановлению речи направлены на развитие словесного мышления, фонации, правильного дыхания, артикуляции, правильной расстановки ударений. Также большое значение уделяется улавливанию высоты звуков, ритма и т.п. Хочу отметить, что все эти упражнения теряют смысл, если ребенку не одеть слуховой аппарат, хотя бы оптимального уровня. Так как научить говорить, когда не слышишь окружающей речи становится практически невозможно и эффект всех занятий без слухопротезирования сводится к нулю. Приобрести ребенку оптимальный или премиум аппарат все равно, что подарить надежду на будущее, так как последние технологии в полном объеме реализуются только в премиум моделях. Чем модель ниже классом, тем меньше в ней адаптационных механизмов и качество звука хуже.
Очень важно предупредить прогрессирование нарушения речи и слуха у маленьких детей. Если годовалый или двухлетний ребенок перестал слышать, скорее всего, он станет немым. Если глухота случилась в возрасте двух-четырех лет, несформированная речь может сохраниться до года. Если ребенок оглох в возрасте пяти-шести лет, его речь станет невыразительной, неразборчивой, монотонной. Поскольку нарушается контакт с окружающими, у ребенка меняется психика.
Родители должны вовремя обследовать и начать лечить такого малыша, а при неэффективности лечения в ближайшие несколько месяцев – одевать слуховой аппарат. В настоящее время существует много средств облегчения жизни глухих детей. Это и компьютерные программы различного назначения, и портативные аппараты искусственной речи, и дешифраторы, воспроизводящие субтитры в телепередачах для глухих, и т.п. Но это все может и не понадобиться, при наличии остатков слуха и грамотном слухопротезировании, периодическим контролем за ношением слуховых аппаратов. Например, в оптимальных и премиум моделях слуховых аппаратов есть бортовой журнал, который постоянно фиксирует, сколько времени ребенок носит слуховой аппарат и в каких шумовых ситуациях он бывает. Это может дать ценную информацию, как помочь данному конкретному ребенку в его ситуации. Шаблонное мышление здесь не применимо, так нарушение слуха у всех разное, восприятие звуков тоже. Чем больше индивидуальный подход, тем выше эффект и лучше помощь ребенку. Слуховые аппараты более высокого класса расширяют возможности индивидуального подхода в решении проблемы плохого слуха.
Дыхательная гимнастика
Заикание и прочие нарушения речи обычно сопровождаются нарушением дыхания, поэтому при лечении большое значение уделяется дыхательной гимнастике. Главные ее аспекты – ритмичность и самовнушение.
Рекомендуется делать следующее упражнение:
Данное упражнение рекомендуется делать 4 раза в день. В дальнейшем можно постепенно увеличивать время вдохов и выдохов до 12 ударов пульса. Внимание: дыхательную гимнастику нельзя делать перед сном. |
глухонемота — это… Что такое глухонемота?
глухонемота — глухонемота … Орфографический словарь-справочник
ГЛУХОНЕМОТА — ГЛУХОНЕМОТА. Содержание: Виды Г………………… 397 Этиология Г………………. 398 Пат. анатомические изменения…….. 398 Остатки стуха и расстройства вестибулярного аппарата при Г……………. 39 9 Диагноз, прогноз и лечение … Большая медицинская энциклопедия
глухонемота — ы; ж. Неспособность к словесной речи вследствие врождённой или приобретённой в раннем детстве глухоты. * * * глухонемота неспособность к словесной речи, обусловленная врождённой или приобретённой в раннем детстве глухотой. В Российской Федерации… … Энциклопедический словарь
ГЛУХОНЕМОТА — ГЛУХОНЕМОТА, глухонемоты, мн. нет, жен. (книжн. спец.). Отсутствие слуха и способности речи. Толковый словарь Ушакова. Д.Н. Ушаков. 1935 1940 … Толковый словарь Ушакова
ГЛУХОНЕМОТА — ГЛУХОНЕМОТА, ы, жен. (спец.). Отсутствие слуха и словесной речи. Толковый словарь Ожегова. С.И. Ожегов, Н.Ю. Шведова. 1949 1992 … Толковый словарь Ожегова
ГЛУХОНЕМОТА — неспособность к словесной речи, обусловленная врожденной или приобретенной в раннем детстве глухотой. В Российской Федерации осуществляется обязательное обучение глухонемых детей в специальных дошкольных учреждениях и школах; они овладевают… … Большой Энциклопедический словарь
Глухонемота — ж. Неспособность к словесной речи вследствие врождённой или приобретенной в раннем детстве глухоты. Толковый словарь Ефремовой. Т. Ф. Ефремова. 2000 … Современный толковый словарь русского языка Ефремовой
глухонемота — глухонемота, глухонемоты, глухонемоты, глухонемот, глухонемоте, глухонемотам, глухонемоту, глухонемоты, глухонемотой, глухонемотою, глухонемотами, глухонемоте, глухонемотах (Источник: «Полная акцентуированная парадигма по А. А. Зализняку») … Формы слов
Глухонемота — В данной статье или разделе имеется список источников или внешних ссылок, но источники отдельных утверждений остаются неясными из за отсутствия сносок … Википедия
Глухонемота — I Глухонемота врожденная или приобретенная в раннем детском возрасте глухота и обусловленное ею отсутствие речи. Развитие речи в норме происходит на основе слухового восприятия речи окружающих и подражания ей. Если ребенок рождается глухим или… … Медицинская энциклопедия
глухонемота — kurčnebylystė statusas T sritis švietimas apibrėžtis Kalbos netekimas dėl įgimto kurtumo ir ankstyvo kalbos centrų sužalojimo. Kurtumas gali būti įvairus. Kurčiu laikomas žmogus, kuris išgirsta tik stipresnį nei 80 decibelų garsą. Kurčnebyliai… … Enciklopedinis edukologijos žodynas
Врожденная глухота
Тимоти К. Хейн, доктор медицины • Прослушивание страницы • Последнее изменение страницы: 16 мая 2021 г.
При приобретенной глухоте, связанной с возрастом или шумовым воздействием примерно на 2 порядка чаще, чем генетическая глухота, врожденная глухота (присутствует с рождения) встречается у 1 на каждые 1000-2000 рождений с аутосомно-рецессивным наследованием самая распространенная форма (более 75%). Примерно 50% этой потери слуха является генетической, 25% — приобретенной и 25% — по неизвестной причине.
Аномалии внутреннего уха, такие как уродство Мондини, с различными моделями наследования, составляют примерно 20% врожденная нейросенсорная глухота. Основная часть оставшейся генетической глухоты является несиндромальным, что означает, что он не имеет каких-либо очевидных отличительных черт.
Большинство этих нарушений задокументировано с помощью генетического картирования. Чтобы это сработало, в семье должно быть более 10 пострадавших членов. Маркерный анализ позволяет идентифицировать область генома, в которой находится ген заболевания.
Совсем недавно секвенирование экзома, по данным Дауни и др. (2020), по состоянию на 2020 год стоит около 1000 долларов на ребенка и дает 30 диагнозов на 1000 протестированных детей. Эти авторы считали, что это было рентабельно, потому что (в Австралии) «средняя готовность общества платить за секвенирование экзома оценивалась в 4600 австралийских долларов на каждого протестированного ребенка по сравнению со стандартным уходом, что дало положительную чистую выгоду в размере 3600 австралийских долларов». Однако мы не совсем уверены, что «общественная готовность» является лучшим сравнением для всех ситуаций.Например, в других странах, помимо Австралии, выделение ограниченных ресурсов здравоохранения на мероприятия, более непосредственно влияющие на здоровье населения, может быть более важным, чем выделение ресурсов на диагностику генетической глухоты.
Среди детей с нейросенсорной тугоухостью любого типа от 11 до 41% из них имеют внутренние аномалии, видимые на КТ. Среди детей, у которых предполагается наличие аномалий внутреннего уха и которых направляют на МРТ, около 40% имеют аномалии (McClay et al, 2008).Конечно, это не означает, что 40% всех глухих детей будут иметь аномалии МРТ, а скорее 40% тех, у кого врачи считают аномалиями МРТ и которых направляют на МРТ, есть аномалии МРТ. Другими словами, это означает, что частота отклонений от нормы для врачей в том месте, где проводилось это исследование (Даллас), составляет около 40%.
Врожденная врожденная глухота
Прежде чем мы начнем говорить об отдельных синдромах, наследственная глухота обычно бывает симметричной и двусторонней, почти всегда нейросенсорной и обычно более тяжелой на высоких частотах.Однако особый паттерн потери слуха, называемый «укусом печенья», обычно предполагает генетический паттерн — другими словами, это довольно специфический признак генетического паттерна глухоты. Приблизительно 2/3 людей с привычкой кусать печенье имели наследственную потерю слуха в исследовании одной академической практики (Shah and Blevins, 2005). Кажется вероятным, что за пределами академической среды паттерны слуха, связанные с укусом печенья, с большей вероятностью связаны с наследственной потерей слуха.
Несиндромальный (80% генетической глухоты):
Около 80% генетической потери слуха не является синдромом. Другими словами, хотя о генетических синдромах гораздо больше статей, чем о несиндромальной глухоте, и на этой странице гораздо больше текста, эти состояния, которые являются предметом стольких дискуссий, являются лишь маленькой частью большого генетического загадка потери слуха.
В период с 1992 по 2001 гг. Картировано 38 локусов аутосомно-доминантной несиндромальной глухоты и 11 гены были клонированы. Аутосомно-доминантные локусы называются DFNA, аутосомными. рецессивный как DFNB и X-связанный как DFN.Обновленную информацию о текущем местоположении можно найти о наследственной тугоухости домашняя страница, размещенная в Университете Айовы. Несиндромальная глухота очень гетерогенный, но мутации в молекуле коннексина-26 (щелевое соединение белок, ген GJB2) составляют около 49% пациентов с несиндромальной глухотой. и около 37% спорадических случаев.
Анализы на коннексин-26 коммерчески доступны. в нескольких лабораториях. Примерно 1 из 31 человека европейской добычи являются вероятными носителями.Однако популяционный анализ показывает, что что существует более 100 генов, участвующих в несиндромальном нарушении слуха (Morton, 1991). Одна мутация особенно распространена, а именно 30delG.
Существует номенклатура несиндромальной глухоты:
Аутосомно-доминантный (DFNA)
Аутосомно-доминантная глухота передается напрямую из поколения в поколение. Это часто можно определить аутосомно-доминантный образец с помощью простого осмотра генеалогического древа.Примерами аутосомно-доминантной глухоты являются миссенс-мутации. в COL11A2 (DFNA13) (Leenheer et al, 2001). COL11A2 кодирует цепочку типа XI коллаген. В качестве примера фенотипа глухоты в DFNA10 наблюдается постлингвальный, изначально прогрессирующий и возникающий без влияния пресбиакузиса, в значительной степени стабильной, плоской нейросенсорной глухоты (De Leenheer et al, 2001).
DFNA9 / COCH может изначально напоминать Меньера болезнь, но она прогрессирует и заканчивается тяжелой глухотой и двусторонним вестибулярным потеря.(Лемэр и др., 2003)
DFNA11 также может вызывать вестибулярные проблемы (Jen, 2009)
Мутация DFNA6 / 14-WFS1 представляет собой прогрессирующую низкочастотную нейросенсорную нарушение слуха (LFSNHL), вызванное гетерозиготной мутацией WFS1. (Пеннингс и др., 2003). Мутации в гене WFS1 являются наиболее распространенной формой доминантного низкочастотная нейросенсорная тугоухость. Дифференциальный диагноз низкого частоты SNHL включают внезапную потерю слуха, FDNA1, DFNA6 / 14, LFSNHL, ассоциированная с болезнью Меньера, и спорадический LFNSHL.
- De Линхеер Э.М., Хюйген П.Л., Уэйн С., Смит Р.Дж., Кремерс С.В. Фенотип DFNA10. Отол Ринол Ларингол 2001 Сентябрь; 110 (9): 861-6
- Pennings RJE и другие. Прогрессирование низкочастотного нейросенсорного слуха потеря (DFNA6 / 14-WFS1). Arch OtoHNS 2003: 129; 421-42
.- Лесперанс М.М. и другие. Мутации в гене синдрома Вольфрама типа I (WFS1) определяют клиническую сущность доминантного низкочастотного нейросенсорного слуха. потеря.Arch Oto HNS 2003: 129: 411-420
Аутосомно-рецессивный (DFNB)
Аутосомно-рецессивным расстройствам требуется ген как от матери, так и от отца.
DFNB1 (коннексин 26) — наиболее распространенная форма генетической потери слуха. Проявляется предъязыковой глухотой, иногда с потерей слуха от легкой до умеренной. Вестибулярных или рентгенологических отклонений нет. Это вызвано мутацией в белке щелевого соединения. В США действует тарифная ставка 3%.
Мутации, связанные с DFNA6 / 14-WFS1, вызывают рецессивный синдром, известный как Синдром Вольфрама с несахарным диабетом, сахарным диабетом, атрофией зрительного нерва и глухота (Lesperance et al, 2003).
Биаллелик
Синдромная глухота (оставшиеся 20% врожденной глухоты)Этот необычный тип генетической проблемы означает, что существует мутация (не обязательно одинаковая) в обеих копиях определенного гена (отцовской и отцовской).
Синдром MEGDEL (3-метилглутаконовая ацидурия, дистония-глухота, энцефалопатия, Leigh-like) обусловлен подвижностью в гене SERAC1, который кодирует белок с серин-липазным доменом.Маас и др. (2017) сообщили об этом редком синдроме в 2017 году. Его следует рассматривать как синдром прогрессирующей глухоты-дистонии с частым поражением печени. Часто встречаются спастичность и дистония. МРТ показывает патогномонический «скорлупный глаз». Дожить до взрослой жизни — обычное дело.
Это чрезвычайно сложная взаимосвязанная совокупность расстройств. Описания здесь только для того, чтобы дать общее представление о болезнях и не предназначены для включать все признаки расстройства.В большинстве случаев ссылка на базу данных OMIM указан основной тип генетического нарушения.
Альпорт синдром — это фактически прогрессирующая двусторонняя потеря слуха, а не врожденная потеря слуха.
Синдром Альпорта вызывается мутациями в COL4A3, COL4A4 или COL4A5. Это гены, влияющие на коллаген. Классический фенотип — почечная недостаточность и прогрессирующая нейросенсорная глухота. Нарушение слуха двустороннее и коррелирует с возрастом (Moon et al, 2009).Ассоциации альпорта и головокружения нет.
Синдром Бараката (текст любезно предоставлен доктором Баракатом).
Синдром Бараката, также известный как синдром HDR, представляет собой клинически гетерогенное, редкое генетическое заболевание, характеризующееся триадой гипопаратиреоза, нейросенсорной глухоты и почечной недостаточности (Barakat et al, 1977; 2020). В большинстве случаев синдром вызван делециями или мутациями фактора транскрипции цинкового пальца GATA3 на хромосоме 10p14. Наследование аутосомно-доминантное.Синдром следует рассматривать, если потеря слуха выявляется при обычном неонатальном тесте слуха или если почечные аномалии обнаруживаются при обычном пренатальном УЗИ. Синдром также может проявляться гипокальциемией, тетанией или афебрильными судорогами в любом возрасте.
Потеря слуха встречается у 96% пациентов и характеризуется ранним началом, умеренной и тяжелой нейросенсорной тугоухостью, обычно двусторонней и несколько хуже на верхнем конце частотного спектра. Наружные волосковые клетки играют важную роль в этиологии потери слуха.Использование панелей генов секвенирования следующего поколения, которые включают GATA3, у пациентов с явно изолированной глухотой, позволило на раннем этапе идентифицировать мутации GATA3 у пациентов с ранее нераспознанным синдромом Бараката. Лечение слуха должно быть назначено детям как можно раньше, чтобы помочь их речи, языку и социальным навыкам полностью раскрыть свой потенциал.
Гипопаратиреоз встречается у 93% пациентов, а различные заболевания почек — у 72%. Ведение состоит из лечения клинических отклонений во время обращения.Прогноз зависит от тяжести заболевания почек.
Branchio-Oto-Renal Синдром
Бранчио-ото-почечный синдром вызывается мутациями в EYA1, гене 16 экзонов. в пределах геномного интервала 156 кБ. Для этого синдрома характерны слуховые нарушениях и катаракте, жаберных свищах и преаурикулярных ямках. Могут возникнуть пороки развития Мондини и связанные с ними дисплазии.
Шарко Мари Зуб (CMT) с соединением X-образной связкой
Доминантно наследуемая форма Х-сцепленного ШМТ вызывается мутацией в Ген коннексина 32, картированный в локусе Xq13.Обычные клинические признаки состоят из периферическая невропатия в сочетании с проблемами стопы и «бутылкой шампанского» телят. У некоторых возникает нейросенсорная глухота. (Стойкович и др., 1999). У некоторых из этих пациентов есть слуховая невропатия.
Как отмечалось выше, ген коннексина также связан с большим процентом случаев несиндромальной глухоты. Есть несколько других связанных невропатий. и синдромы глухоты. Аутосомно-рецессивная демиелинизирующая нейропатия, аутосомная доминантные наследственные невропатии I и II типа и Х-сцепленные наследственные аксональные невропатии с умственной отсталостью связаны с глухотой (Стойкович и др., 1999).
Болезнь Фабри
Болезнь Фабри (БФ) — это Х-сцепленное рецессивное наследственное лизосомное нарушение накопления, которое приводит к накоплению глоботриаозилцерамида (Gb3) в тканях почек и сердца, а также в центральной и периферической нервной системе. По данным Koping et al (2017), «сенсоневральная потеря слуха была обнаружена в 58,8% когорты, которая обычно возникала во внезапных эпизодах и затрагивала особенно высокие частоты. Потеря слуха асимметрична, начинается односторонне и позже поражает контралатеральное ухо.О тиннитусе сообщили 41,2% «.
- Köping M1, Shehata-Dieler W1, Cebulla M1, Rak K1, Oder D2, Müntze J2, Nordbeck P2, Wanner C2, Hagen R1, Schraven S1 Дисфункция сердца и почек связана с прогрессирующей потерей слуха у пациентов с болезнью Фабри. .PLoS One. 2017 21 ноября; 12 (11): e0188103. DOI: 10.1371 / journal.pone.0188103. eCollection 2017.
Синдром Гольденхара.
Окулоаурикуловертебральная дисплазия (OAVD) или синдром Гольденхара первоначально была описан в 1881 г.Он включает в себя комплекс признаков, включая гемифациальную микротию, отомандибарный дизостоз, эпибульбарные липодермоиды, колобома и позвоночные аномалии которые происходят из-за сосудистых и генетических аберраций развития. Она имеет разнообразная этиология и не относится к одному генетическому локусу. Заболеваемость составляет примерно 1 из 45 000. (Scholtz et al, 2001).
Джервелл и синдром Ланге-Нильсена
Этот синдром слуха связан с сердечной аритмией.Есть продление интервала QT, аритмии torsade de pointe (поворот точек, в ссылка на очевидное чередование положительных и отрицательных комплексов QRS), внезапные обмороки и тяжелая или глубокая сенсоневральная потеря слуха.
Klippel-Feil.
Клиппель-Фейл (KFS) — врожденная аномалия шейных (шейных) позвонков. Проявляется короткой шеей, заниженной линией волос и ограниченной подвижностью шеи. Это связано с врожденными аномалиями всех трех частей уха (наружного, среднего и внутреннего уха), а также IAC и вестибулярного водопровода (см. Ниже).По данным Yildirim et al (2008), около 60% пациентов с KFS имеют аномалии уха. Корреляции между патологией уха и аномалиями скелета или вне скелета не было.
- Йилдирим Н., Арсланоглу А., Махирогуллари М., Сахан М., Озкан Х. Синдром Клиппель-Фейля и связанные с ним аномалии уха. Ам Дж. Отоларингол. 2008 сентябрь-октябрь; 29 (5): 319-25.
Синдром большого вестибулярного акведука (LVAS), также известный как EVA (увеличенный вестибулярный акведук). Хотя это присутствует с рождения, оно также прогрессирует.
При «синдроме большого вестибулярного акведука» наблюдается увеличение эндолимфатического протока (ED на рисунке выше), который соединяет эндолимфатический отсек (синий вверху) с эндолимфатическим мешком (который лежит непосредственно под твердой мозговой оболочкой задней черепной ямки, ES вверху) . См. Страницу EVA об этом условии.
Mohr-Tranebjaerg синдром (DFN-1)
Синдром Мора-Транебьерга (DFN-1) — это Х-сцепленный рецессивный синдромальный слух. потеря, характеризующаяся постлингвальной сенсоневральной глухотой в детстве, за которой последовала прогрессирующей дистонией, спастичностью, дисфагией и атрофией зрительного нерва.Синдром вызвано мутацией, которая, как считается, приводит к дисфункции митохондрий. Это похоже на спиноцеребеллярная дегенерация, называемая Фредрейховой атаксия, которая также может проявляться нейросенсорной тугоухостью, атаксией и оптическим атрофия. Кардиомиопатия, характерная для Freidreichs, не наблюдается в Mohr-Tranebjaerg.
Норри Болезнь.
Классические признаки включают специфические глазные симптомы (псевдоопухоль сетчатки, гиперплазия сетчатки, гипоплазия и некроз внутреннего слоя сетчатки, катаракта, луковичный туберкулез), прогрессирующая нейросенсорная тугоухость и психическая нарушение слуха, хотя менее половины пациентов страдают нарушениями слуха или умственно отсталый.
Синдром Окихиро
Классические признаки включают синдром Дуэйна (напоминает паралич 6-го нерва), врожденную гипоплазию зрительного нерва, двустороннюю глухоту и пороки развития лучевого луча. Это связано с другими нарушениями SALL4, включая акро-почечно-глазной синдром и синдром Холта-Орама. Согласно Chun et al., Нейросенсорная глухота встречалась только у 17% из 41 пациента (2001).
Пендред Синдром
Синдром Пендреда — одна из наиболее частых синдромальных форм глухоты.По сути, это глухота в сочетании с заболеванием щитовидной железы (эутиреоидный зоб) (Wemeau, Kopp, 2017). Вестибулярный тестирование, особенно ротационное тестирование, если таковое имеется, должно проводиться в случаи с известными мутациями. Это происходит из-за мутации переносчика сульфат-иона 7q31. Это аутосомно-рецессивное заболевание. По состоянию на 2006 год было зарегистрировано 90 мутаций в этом гене (Cho et al, 2006). Пендред связан с синдромом большого вестибулярного водопровода (см. Выше), а также с синдромом Мондини (см. Ниже). Обратите внимание, что многие люди с проблемами щитовидной железы страдают болезнью Меньера (Brenner et al, 2004), и, таким образом, LVAS, синдром Меньера и синдром Пендреда могут быть взаимосвязаны.
Около 60% мутаций в гене SLC26A4, которые, как известно, вызывают синдром Пендреда, могут быть обнаружены с помощью генетического тестирования. Это вариант для людей с соответствующими симптомами или рентгенологическими заболеваниями.
Мы наблюдали очень мало пациентов с синдромом Пендреда в Чикаго. Головокружение и слух, что позволяет предположить, что это нечастая причина головокружения в целом. Мы столкнулись с одной семьей с Пендредом у одного человека и со многими другими людьми без проблем со слухом, имеющими головокружение типа ДППГ, что позволяет предположить, что у носителей головокружения может быть некоторый риск.В литературе это не обсуждается.
Спинальная мышечная атрофия (СМА) и симптомы слуха.
Хотя SMA обычно не связаны с симптомами слуха, недавний отчет предполагает, что нарушение, вызванное мутацией в TRPV4, может вызывать невропатию, а также потерю слуха (Oonk et al, 2014).
Наклейка синдром.
Мутации в COL11 являются причиной синдрома Стиклера.
Синдром Стиклера наследуется по аутосомно-доминантному типу.Клинические признаки включают врожденную катаракту, тяжелую миопию, черепно-лицевые особенности, включая гипоплазию средней зоны лица, раздвоение язычка, волчью пасть и последовательность Пьера Робена. Нейросенсорная тугоухость, гипермобильные суставы и преждевременный остеоартрит также являются общими признаками. Для этого синдрома характерно прогрессирующей миопией в первый год жизни и артропатии.
Предатель Синдром Коллинза (OMIM Entry TCOF1)
Синдром Тричера Коллинза характеризуется колобомой нижнего века. (при синдроме Гольденхара задействовано верхнее веко), микрогнатия, микротия, гипоплазия скуловых дуг, макростомия и нижнее смещение латеральные уголки глаз по отношению к медиальным уголкам глаз.
Синдром Тернера.
Синдром Тернера встречается примерно у 1/2000 женских родов. Большинство людей с синдромом Тернера имеют только одну копию Х-хромосомы и не имеют Y. Примерно две трети населения Тернера страдают потерей слуха, что примерно поровну разделено между нейросенсорным и проводящим типами (Ingeborg et al, 2005).
Waardenburg синдромы типа I и II
Синдром Ваарденбурга (WS) — это преимущественно аутосомно-доминантное заболевание, характеризующееся пигментными аномалиями кожи, волос, глаз и различными дефектами других тканей, происходящих от нервного гребня (Read and Newton, 1997).На его долю приходится более 2% врожденных нарушений слуха. На основании клинических и генетических критериев распознаются по крайней мере четыре типа. (Апайдин и др., 2004). Расстройство не очень однородно, даже в пределах одной семьи. Около 1/30 человек в школах для глухих страдают синдромом Ваарденбурга.
Существует четыре подтипа WS. WS1 в основном вызывается мутациями PAX3, в то время как мутации MITF, SNAI2 и SOX10 связаны с WS2. Сообщается о более чем 100 различных болезнетворных мутациях во многих этнических группах (Chen et al, 2010).Мутация SOX10 также участвует в синдроме Каллмана с глухотой (Pingault et al, 2015). MITF (фактор транскрипции микрофтальмии) связан с процессом меланогенеза (т. Е. Пигментом), и мутации также могут приводить к синдрому Титца (Otreba et al, 2012).
Клинические признаки синдрома Ваарденбурга (WS) включают боковое смещение внутреннего угла глазной щели каждого глаза, пигментные аномалии волос, радужной оболочки и кожа (часто белый чуб и гетерохромия радужки — см. выше; или светло-голубые глаза) и нейросенсорная глухота.У некоторых пациентов также наблюдается головокружение (Black et al, 2001). Смещение угла глазной щели, dystropia cantorum, является отличительной чертой между WS1 (есть) и WS2 (нет). Сочетание характеристик WS I типа с аномалиями верхних конечностей имеет был назван синдромом Клейна-Ваарденбурга или WS типа III. Сочетание рецессивно унаследованные характеристики WS типа II с болезнью Гиршпрунга получили название Синдром Ваарденбурга-Шаха или WS IV типа. Мутации генов EDNRB, EDN3 и SOX10 ответственны за синдром Ваарденбурга типа IV (Otreba et al, 2013).
Ushers синдром. — наиболее частая причина одновременной глухоты и слепоты
Синдром Ушера характеризуется нарушением слуха и пигментным ретинитом (Young, Mets and Hain; 1996). Синдром Ушера можно разделить на 3 различных типа на основе клинических проявлений. Выводы.
В последние годы было обнаружено, что гены Ашера довольно распространены — примерно 1/70 человек имеет единственную мутацию.
При первом типе наблюдается нарушение слуха и вестибулярного аппарата.
При типе II имеется нарушение слуха без вестибулярного нарушения. Однако есть некоторые разногласия по этому поводу, поскольку некоторые хорошо задокументированные пациенты USH-2 имеют ненормальное тестирование, включая VHIT, и уменьшенное или отсутствующее тестирование VEMP (Magliulo et al, 2017). Мы немного сомневаемся в этом, поскольку тесты, в которых были выявлены отклонения от нормы, часто являются отклонениями от нормы в нормальной популяции. Тем не менее, кажется, стоит изучить этот вопрос подробнее.
В типе в-третьих, имеется различная степень вестибулярного нарушения.Пациенты Ushers могут получить пользу от кохлеарного имплантата. Электроретинограмма обычно требуется для получения четкого диагноза (Loundon et al, 2003). Вестибулярный Если возможно, тестирование следует провести в Usher’s.
Митохондриальные нарушения.
См. Эту страницу для отдельного обсуждения.
Врожденная глухота с вариабельной наследственностью
Синдром Вольфрама, впервые описанный в 1938 году, также известен как DIDMOAD (несахарный диабет, сахарный диабет, атрофия зрительного нерва, глухота).Согласно Плантинге и др. (2008), все пациенты имели сходную нейросенсорную тугоухость с плавным понижением слуха. С возрастом не прогрессирует.
Плантинга и др. (2008). Нарушение слуха при генотипированных патиниях с синдромом Вольфрама. Ann ORL 117 (7) 474-500
Врожденная глухота без наследства
На эти типы аномалий приходится примерно 20% врожденной глухоты, остальные имеют генетическое происхождение. Как правило, эти расстройства могут быть связаны с генетическими нарушениями, но чаще возникают самостоятельно.
Вирусные синдромы
Врожденную потерю слуха часто связывают с пренатальными инфекциями с нейротрофическим вирусы, такие как корь или цитомегаловирус (ЦМВ). Недавнее исследование показало, что «более 40% глухих по неизвестной причине, нуждающихся в реабилитации» относится к ЦМВ. (Барби и др., 2003). ЦМВ — самая распространенная внутриутробная инфекция в США. Младенцы могут контактировать с грудным молоком. Другие жидкости организма также могут передавать ЦМВ (например,г. моча, слюна). В развитых странах пожилые люди подвергаются воздействию вторичных механизмов.
Отсроченное начало потери слуха является обычным явлением — младенцы с ЦМВ и нормальным слухом при рождении должны находиться под наблюдением в течение 6 лет. Новорожденных с ЦМВ можно лечить ганцикловиром. Это лечение необходимо очень тщательно контролировать, так как у 2/3 младенцев развивается нейтропения.
| С деформированной улиткой | Потеря слуха |
| глубокий |
| Тяжелая | |
| глубокий |
| Умеренная, переменная | |
| От умеренной до тяжелой (сохраняются более высокие частоты) |
| С нормальной улиткой | |
| мягкий |
| От умеренной до тяжелой (ступенчато) |
| (Изменено из Джеклера и др.) | |
Врожденные пороки развития цепи слуховых косточек.
Эти пороки развития вызывают кондуктивную тугоухость. Пороки развития класса III по Тойниссену и Кремеру являются примерами подвижной ступни стремени. Как и в случае с большинством заболеваний среднего уха, их часто можно лечить хирургическим путем. (Винсент и др., 2016)
Пороки развития бокового полукружного канала
Согласно Venkatasamy et al (2019), «пороки развития LSCC обычно связаны с потерей слуха (61%), особенно SHNL (39%).«Можно было бы ожидать, что у этих пациентов также будет вестибулярная дисфункция.
Дисплазия Мондини и Мишеля (дополнительную информацию см. На странице Мондини)
КТ височной кости обычно проводится людям с нейросенсорной потеря слуха. Около 25% пациентов с врожденной тугоухостью имеют костный пороки развития внутреннего уха (Mafong et al, 2002).
Нормальная улитка имеет два с половиной оборота. Улитковая аномалия состоит из мембранозной аномалии, костной аномалии или их комбинации два.Если развитие улитки у эмбриона остановлено, общая полость может возникают вместо улитки, как улитка. Полный лабиринт и улитка аплазия называется деформацией Мишеля (см. рисунок справа от Strome).
Неполный раздел называется Дисплазия или порок развития Мондини. Кроме того, он состоит из кистозной вершины, расширенный вестибюль и большой вестибулярный водопровод.
Пациенты с общим синдромом Дауна (трисомия 21) часто имеют пороки развития внутреннего уха.
Деформации перепончатого лабиринта — Шибе и Александр
Есть также некоторые деформации перепончатого лабиринта — например, очень распространенная деформация Шибе (pars inferior — улитка и мешочек).
Аплазия Александра характеризуется аплазией протока улитки. Наиболее заметно поражается кортиев орган, особенно базальный поворот улитки и прилегающие ганглиозные клетки. Потеря слуха наиболее заметна на высоких частотах, в то время как низкочастотный слух относительно сохраняется.
Частота этих заболеваний в основном обусловлена вскрытием височной кости. Причина в том, что эти деформации не могут быть диагностированы с помощью компьютерной томографии, так как компьютерная томография не может определить аномалии перепончатого лабиринта. Для визуализации этих структур использовалась МРТ высокого разрешения. Практически однако обычные МРТ-сканеры 1,5 тесла не предоставить достаточно подробностей, чтобы иметь большую клиническую ценность. Более новые сканеры 3.0 тесла могут быть более ценными.
Можно было бы подумать, что тест VEMP будет хорошим методом обнаружения деформации Шибе, поскольку VEMP чувствителен к нарушениям мешочка.
Пороки развития внутреннего слухового прохода.
Отверстия необычного размера между внутренним ухом и мозгом (внутренний слуховой проход) обычно связаны с другими костными аномалиями (сюрприз!). Ли и др. (2014).
Артикул:
- Apaydin, F., et al. (2004). «[Синдром Ваарденбурга. Гетерогенное заболевание с переменной пенетрантностью]». HNO 52 (6): 533-537.
- Barakat, AY, D’Albora, JB, Martin, MM, Jose, PA.Семейный нефроз, нервная глухота и гипопаратиреоз. J. Pediat 1977; 91: 61-4. Онлайн-Менделирующее наследование в человеке, Университет Джона Хопкинса № 146255.
- Баракат А.Дж., Заизал Х. Характеристики потери слуха при синдроме Баракат. Энн Пед Рес, 2020; 4 выпуск 5, статья 1051 (открытый доступ)
- Barbi M, Binda S, Caroppo S, Ambrosetti U, Corbetta C, Sergi P. Более широкий роль врожденной цитомегаловирусной инфекции в нейросенсорной тугоухости. Pediatr Infect Dis J, 2003, январь; 22 (1): 39-42
- Черный, F.O., et al. (2001). «Вестибулярный фенотип синдрома Ваарденбурга?» Отол Нейротол 22 (2): 188-194.
- Brenner M, Hoistad D, Hain TC. Распространенность дисфункции щитовидной железы при болезни Меньера. Архив Oto HNS, 130/2 226-228 (2004)
- Cho, M.A., et al. (2006). «Мутация H723R в гене PDS / SLC26A4 связана с типичным синдромом Пендреда у корейских пациентов». Эндокринная 30 (2): 237-243.
- Chun, B. B., et al. (2001). «Характеристики синдрома Окихиро.»J Pediatr Ophthalmol Strabismus 38 (4): 235-239.
- Chen, H., et al. (2010). «Новые мутации генов PAX3, MITF и SOX10 у китайских пациентов с синдромом Ваарденбурга I или II типа». Biochem Biophys Res Commun 397 (1): 70-74.
- Chen K, Wang X, Sun L., Jiang H. Скрининг SLC26A4, FOXI1, KCNJ10 и GJB2 у пациентов с двусторонней глухотой с пороками развития внутреннего уха. Otolaryngol Head Neck Surg. 2012 г. 12 марта [Epub перед печатью]
- Лилиан Дауни, Дэвид Джей Амор, Джейн Холлидей, Шэрон Льюис, Мелисса Мартин, Илиас Горанитис.Секвенирование экзома для изолированной врожденной потери слуха: анализ экономической эффективности. Ларингоскоп. 31 декабря 2020 г. doi: 10.1002 / lary.29356. Интернет впереди печати.
- Эль-Шахави М и другие. Две большие испанские родословные с несиндромными нейросенсорная глухота и мутация мтДНК в нуклеотиде 1555 в гене 12S рРНК. Доказательства гетероплазмии. Неврология 1997; 48: 453- .
- Ингеборг Ю.М. и другие. Отологические заболевания при синдроме Тернера. Отология и невротология 26: 145-150, 2005
- Джен, Дж.С. (2009). «Двусторонняя вестибулопатия: клинические, диагностические и генетические соображения». Semin Neurol 29 (5): 528-533.
- Leenheer и другие. Аутосомно-доминантное наследственное нарушение слуха, вызванное миссенс-мутацией в COLA11A2 (DFNA13).
- Lemaire FX, Feenstra L, Huygen PL, Fransen E, Devriendt K, Van Camp G, Vantrappen Дж., Кремер С. В., Ваким П. А. и Косс Дж. К. (2003). «Прогрессивная нейросенсорная потеря слуха и вестибулярное нарушение с головокружением (DFNA9 / COCH): продольное анализы в бельгийской семье.«Отол Нейротол 24 (5): 743-8.
- Li Y1, Yang J, Liu J, Wu H. Исследование пороков развития внутреннего слухового прохода, канала улиткового нерва и улиткового нерва. Eur Arch Otorhinolaryngol. 2014 6 марта [Epub перед печатью]
- Loundon N и другие. Синдром Ушера и кохлеарная имплантация. Отол Нейротол 24: 216-221, 2003
- Maas. Прогрессирующая глухота-дистония из-за мутаций SERAC1: исследование 67 случаев. Энн Нейрол 2017: 82: 1004-1015
- Mafong DD и другие.Использование лабораторной оценки и радиологической визуализации в диагностической оценке детей с нейросенсорной тугоухостью. Ларингоскоп 2002: 112: 1-7
- Magliulo G, Iannella G, Gagliardi S, Iozzo N, Plateroti R, Mariottini A, Torricelli F. Синдром Ашера типа II: сравнительное исследование генетических мутаций и оценка вестибулярной системы. .Отоларингол Head Neck Surg. 2017 ноя; 157 (5): 853-860. DOI: 10.1177 / 0194599817715235. Epub 2017 27 июня.
- McClay JE и другие.Оценка нейросенсорной тугоухости у детей с помощью магнитно-резонансной томографии. Arch ORL 2008: 134 (9) 945-952
- Торговец С.Н. и другие. Гистопатологические и генетические исследования височной кости при синдроме Мора-Транебьярта (DFN-1). Отол Нейротол 22: 506-511, 2001
- Moon, I. S., et al. (2009). «Тяжелая или глубокая потеря слуха у пациентов с прогрессирующим синдромом Альпорта». Acta Otolaryngol 129 (9): 982-987.
- Morton NE. 1991. Генетическая эпидемиология нарушения слуха.Энн NYAS 630; 16-31.
- Накамура Ю. и другие. Аномальные вызванные потенциалы синдрома Кернса-Сейра. Electromyogr Clin Neurophysiol 1995: 35: 365-370
- Oonk AM1, Ekker MS2, Huygen PL3, Kunst HP3, Kremer h5, Schelhaas JJ5, Pennings RJ3.] Внутрисемейная переменная потеря слуха при спинальной мышечной атрофии, индуцированной TRPV4. Анн Отол Ринол Ларингол. 2014 июн 24, pii: 0003489414539130.
- Otreba, M., et al. (2012). «[Регуляция меланогенеза: роль цАМФ и MITF].»Postepy Hig Med Dosw (Online) 66: 33-40.
- Otreba, M., et al. (2013). «[Наследственные гипомеланоцитозы: роль генов PAX3, SOX10, MITF, SNAI2, KIT, EDN3 и EDNRB]». Postepy Hig Med Dosw (Online) 67: 1109-1118.
- Pingault, V., et al. (2015). «Мутации SOX10 имитируют изолированную потерю слуха». Clin Genet 88 (4): 352-359.
- Рид, А. П. и В. Э. Ньютон (1997). «Синдром Ваарденбурга». J Med Genet 34 (8): 656-665.
- Scholtz et al. Синдром Голдендара: врожденная кондуктивная недостаточность слуха. или нейросенсорного происхождения? Отология Нейротол 22: 501-505, 2001
- Sennaroglu L, Saatci I.Новая классификация кохлеовестибулярных пороков развития. Ларингоскоп 112: 2230-2241, 2002
- Шах, Р. К., Н. Х. Блевинс и др. (2005). «Среднечастотная нейросенсорная тугоухость: этиология и прогноз». Дж Ларингол Отол 119 (7): 529-33.
- Сталь КП. Новая эра в генетике глухоты. NEJM 1998
- Стойкович и другие. Сенсорно-невральная дефанция при Х-сцепленном соединении Шарко-Мари-Зуба болезнь с мутацией коннексина 32 (R142Q). Неврология 1999: 52: 1010-1014
- Strome SE, Baker KB, Langman AW.Случай месяца для визуализации: Порок развития внутреннего уха. American J. Otology, 19: 396-397, 1998
- Винсент Р., Вегенер И., Деркс Л.С., Грольман В. Врожденные пороки развития слуховых косточек в подвижных стремени у детей: результаты в 17 случаях. Ларингоскоп. 2016 Март; 126 (3): 682-8. DOI: 10.1002 / lary.25351. Epub 2015 23 декабря
- Янг Н.М., Мец МБ, Хайн ТЦ. Ранняя диагностика синдрома Ашера у младенцев и детей. Am J Otol. 1996; 17 (1): 30-4.
- Wiener-Vacher SR, Denise P, Narcey P, Manach Y.Вестибулярная функция у детей с ассоциацией ЗАРЯД. Арка Отоларингол HNS 1999: 125: 342-34
- Вемо, Дж. Л. и П. Копп (2017). «Синдром Пендреда». Лучшая практика Res Clin Endocrinol Metab 31 (2): 213-224.
- Venkatasamy A, Foll DL, Eyermann C, Vuong H, Rohmer D, Charpiot A, Veillon F. Деформации бокового полукружного канала коррелировали с данными аудиограммы. . Eur Arch Otorhinolaryngol. 2019 Апрель; 276 (4): 1029-1034. DOI: 10.1007 / s00405-019-05294-у. Epub 2019 6 февраля
- Ямасоба и другие.Гистопатология улитки, связанная с митохондриальной мутация гена переносящей РНК (leu-UUR). Неврология 1999: 52; 1705-1707
См. Также: //raisingdeafkids.org/»>raisingdeafkids.org
Потеря слуха и ваш ребенок
Что такое потеря слуха?
Потеря слуха может произойти, если какая-либо часть уха не работает обычным образом. Он может варьироваться от легкого до сильного:
- Мягкий: Вы можете слышать некоторые звуки речи, но тихие звуки трудно услышать.
- Умеренный: вы не можете слышать очень много звуков речи, когда кто-то говорит на нормальном уровне.
- Тяжелая: вы не слышите звуки речи, когда кто-то говорит на нормальном уровне. Вы можете слышать только громкие звуки.
- Глубокий: вы не слышите звуки речи. Вы можете слышать только очень громкие звуки.
Потеря слуха — распространенный врожденный дефект. Врожденные дефекты — это структурные изменения, присутствующие при рождении, которые могут повлиять практически на любую часть тела. Они могут повлиять на то, как тело выглядит, работает или и то, и другое.Врожденные дефекты могут вызывать проблемы с общим здоровьем, развитием или работой организма. Каждый год в США до 3 из 1000 детей (менее 1 процента) рождаются с той или иной потерей слуха.
Когда ребенок рождается с потерей слуха, это называется врожденной потерей слуха. Потеря слуха также может развиться позже у младенцев, в детстве или во взрослом возрасте.
Каковы признаки потери слуха?
Признаки потери слуха у вашего ребенка могут включать:
- Не пугаться громких звуков
- Не поворачивается к звуку после 6 месяцев
- Не произносить ни одного слова, например, «мама» или «дада», к тому времени, когда ему исполнится 1 год
- Поворачивает голову, если видит вас, но не, если вы только называете его имя
- Кажется, одни звуки слышны, а другие нет
Если у вашего ребенка в любое время проявляются признаки потери слуха, позвоните его врачу, чтобы проверить слух вашего ребенка.
Какие бывают распространенные типы потери слуха?
Слуховая система вашего ребенка — это система в организме, которая помогает ему слышать. Он придает смысл звуковой информации, передаваемой от уха к мозгу. Проблемы в этих частях слуховой системы могут вызвать потерю слуха:
- Наружное ухо. Это включает часть уха на внешней стороне головы, слуховой проход и внешнюю сторону барабанной перепонки. Барабанная перепонка разделяет внешнее и среднее ухо.
- Среднее ухо. Состоит из внутренней части барабанной перепонки и трех маленьких косточек, называемых косточками. Звук, поступающий в ухо, проходит через слуховой проход к барабанной перепонке, вызывая вибрацию барабанной перепонки (быстрое движение вперед и назад). Когда барабанная перепонка вибрирует, она перемещает косточки. Это помогает звуку перемещаться во внутреннее ухо.
- Внутреннее ухо. Он состоит из улитки (изогнутой трубки, наполненной жидкостью) и каналов, которые помогают поддерживать равновесие. Во внутреннем ухе также есть нервы, которые преобразуют звуковые колебания в сигналы, которые поступают в мозг через слуховой нерв (также называемый слуховым нервом).Слуховой нерв передает звуковую информацию от уха к мозгу.
Общие типы потери слуха включают:
- Кондуктивная тугоухость. Это происходит, когда в наружном или среднем ухе есть проблема, которая замедляет или препятствует прохождению звуковых волн. Проблемы могут включать закупорку слухового прохода или жидкость в среднем ухе. Такая потеря слуха часто носит временный характер и обычно поддается лечению с помощью лекарств или хирургического вмешательства.
- Нейросенсорная тугоухость. Это происходит, когда возникает проблема с работой внутреннего уха или слухового нерва. Это может произойти при повреждении определенных клеток внутреннего уха. Такая потеря слуха обычно необратима.
- Смешанная потеря слуха. Это когда ребенок страдает кондуктивной и нейросенсорной тугоухостью.
- Расстройство спектра слуховой нейропатии (также называемое ANSD). В этом состоянии проблема с внутренним ухом или слуховым нервом не позволяет мозгу воспринимать звук.
Как узнать, что у вашего ребенка потеря слуха?
Центры по контролю и профилактике заболеваний (также называемые CDC) и Американская академия педиатрии (также называемая AAP) рекомендуют всем младенцам проходить обследование на предмет потери слуха до достижения ими 1-месячного возраста. У большинства младенцев проверяют слух в рамках скрининга новорожденных перед выпиской из больницы после рождения. Скрининг новорожденных позволяет выявить серьезные, но редкие и поддающиеся лечению состояния при рождении. Он включает в себя проверку крови, слуха и сердца.
Если ваш ребенок не прошел проверку слуха новорожденного, это не всегда означает, что у него потеря слуха. Но ей нужно пройти полную проверку слуха как можно скорее, до того, как ей исполнится 3 месяца. Полный тест слуха может помочь лечащему врачу вашего ребенка диагностировать потерю слуха.
Если у вашего ребенка потеря слуха, важно немедленно начать лечение. В каждом штате есть программа раннего обнаружения и вмешательства по поводу слуха (также называемая EHDI), которая помогает детям с потерей слуха и их семьям.Он может помочь с полными проверками слуха и другими услугами для вашего ребенка. Вы можете найти свою местную программу EHDI на веб-сайте Национального центра оценки и управления слухом.
Какие проверки слуха являются частью скрининга новорожденных?
Ваш ребенок прошел один из следующих тестов слуха в рамках скрининга новорожденных:
- Слуховая реакция ствола мозга (также называемая ABR или тестом на вызванную слуховую реакцию ствола мозга или BAER). В этом тесте используются пластыри, называемые электродами, и компьютер, чтобы проверить, как мозг и слуховой нерв реагируют на звук.Поставщик вашего ребенка надевает пластырь на голову ребенка и мягкие наушники в уши ребенка. Провайдер отправляет звуки через наушники и измеряет мозговые волны вашего ребенка, чтобы увидеть, как на него реагирует мозг. Этот тест может показать, не получает ли мозг звуковую информацию в ясной форме. Ваш ребенок может спать для этого теста.
- Отоакустическая эмиссия (также называемая OAE). Этот тест проверяет, как внутреннее ухо реагирует на звук. Поставщик вашего ребенка вставляет небольшой наушник в ухо вашего ребенка.Наушник подключен к компьютеру. Наушники воспроизводят звуки, которые должны эхом отражаться в слуховом проходе ребенка. Если эха нет, возможно, у вашего ребенка потеря слуха. Ваш ребенок может спать для этого теста.
Какие еще виды проверки слуха могут проходить младенцы?
Если ваш ребенок не сдает тест ABR или OAE при скрининге новорожденных, ее поставщик направляет ее к аудиологу. Это человек, прошедший специальную подготовку по диагностике и лечению потери слуха у новорожденных, детей и взрослых.Лечащий врач вашего ребенка также может направить вашего ребенка к отоларингологу (также называемому ЛОР). Это врач со специальной подготовкой по уходу за ушами, носом и горлом.
Аудиолог проведет у вашего ребенка полную проверку слуха, чтобы проверить, нет ли потери слуха. Это может включать тесты ABR и OAE, а также другие тесты слуха. Аудиолог также может использовать поведенческую аудиометрию, чтобы проверить, как работают все части уха вашего ребенка. Для этого теста аудиолог видит, как ваш ребенок реагирует на звук, наблюдая за изменениями в поведении.Для этого теста ваш ребенок должен бодрствовать. Аудиолог воспроизводит звук и проверяет, реагирует ли ваш ребенок. Например, глаза вашего ребенка могут шевелиться, или он может повернуть голову, сосать соску, замолчать или прислушаться к звуку. Если ваш ребенок реагирует на звук, аудиолог вручает ему награду, как игрушку с мигалкой. Это называется аудиометрией визуального подкрепления.
Как потеря слуха может повлиять на вашего ребенка?
Раннее обследование, диагностика и лечение могут помочь детям с потерей слуха развить речь, язык и социальные навыки.Без своевременного лечения потеря слуха может привести к:
- Задержка или ограниченное развитие речи и языка. Например, младенцы и дети с потерей слуха могут иметь проблемы с пониманием того, что говорят другие люди, заучиванием новых слов и правильным произношением слов. Дети с нелеченной потерей слуха могут иметь плохие коммуникативные навыки.
- Обучение и социальные проблемы. Без раннего лечения дети с потерей слуха могут иметь проблемы с обучением в школе.Потеря слуха также может затруднить общение с другими детьми.
Что вызывает потерю слуха?
Мы не уверены, что вызывает все формы потери слуха. Возможные причины включают:
Гены. Гены — это части клеток вашего тела, в которых хранятся инструкции о том, как ваше тело растет и работает. Гены передаются от родителей к детям. Гены могут играть роль примерно в половине случаев потери слуха у младенцев и детей. Если у вас или вашего партнера есть семейная история потери слуха, вы можете поговорить с генетическим консультантом, прежде чем забеременеть.Этот человек обучен помочь вам понять, как гены, врожденные дефекты и другие заболевания передаются в семье и как они могут повлиять на ваше здоровье и здоровье вашего ребенка.
Иногда инструкции в генах меняются. Это называется изменением гена или мутацией. Изменения генов могут вызвать два типа потери слуха:
- Синдромный. Потеря слуха возникает в связи с другими проблемами со здоровьем, например, слепотой.
- Несиндромальный. Это когда потеря слуха — единственное заболевание ребенка.Примерно 7 из 10 мутаций, вызывающих потерю слуха (70 процентов), не являются синдромальными.
Преждевременные роды или Низкий вес при рождении . Преждевременные роды — это слишком ранние роды, до 37 недель беременности. Недоношенные дети часто имеют больше проблем со здоровьем (например, потеря слуха) при рождении и в более позднем возрасте, чем доношенные дети. Низкий вес при рождении — это когда ребенок рождается с весом менее 5 фунтов 8 унций. Узнайте больше о том, что вы можете сделать, чтобы снизить риск преждевременных родов.
Проблемы с развитием ушей, головы или лица. Если у вашего ребенка врожденный дефект, который меняет форму или структуру его ушей, головы или лица, у него могут быть проблемы со слухом.
Вирусы и инфекции при беременности. Вы можете передать ребенку определенные вирусы и инфекции во время беременности, которые могут вызвать потерю слуха у него. К ним относятся:
- Цитомегаловирус (также называемый ЦМВ). Это распространенный вирус, вызывающий боль в горле, лихорадку, опухшие железы и усталость (постоянное чувство усталости).
- Герпес. Это инфекция, вызванная вирусом простого герпеса (также называемым ВПГ). Инфекции герпеса могут поражать многие части тела, включая глаза, рот, кожу, ягодицы и гениталии.
- Корь. Корь — это легко передающаяся инфекция. Это может вызвать сыпь, кашель и жар.
- Краснуха (также называемая немецкой корью). Это инфекция, которая вызывает легкие симптомы гриппа и сыпь на коже.
- Сифилис. Это инфекция, передающаяся половым путем (также называемая ИППП), которая сначала вызывает язвы и сыпь.ИППП — это инфекция, которую вы можете получить в результате незащищенного секса или интимного физического контакта с инфицированным человеком. Это означает, что вы заразились им в результате незащищенного секса с человеком, инфицированным сифилисом. Вы также можете заразиться сифилисом при непосредственном контакте (прикосновении или поцелуях) к сифилисовой язве инфицированного человека. Если у вас сифилис во время беременности и вы не лечитесь, это может вызвать серьезные проблемы для вашего ребенка, в том числе потерю слуха.
- Токсоплазмоз. Это инфекция, которую можно заразить от недоваренного мяса или прикосновения к кошачьим фекалам.Это может вызвать такие проблемы, как ломота в теле, головная боль, утомляемость (сильная усталость) или жар.
Инфекции, которые есть у вашего ребенка после рождения. Инфекции, которые могут вызвать потерю слуха, включают:
- Инфекции уха. Чаще всего они вызывают временную потерю слуха. Но если инфекция повреждает барабанную перепонку, кости уха или слуховой нерв вашего ребенка, потеря слуха может быть необратимой. Это редко.
- Другие инфекции, например менингит и корь. Чтобы защитить вашего ребенка от подобных инфекций, убедитесь, что он получил все прививки.Воспользуйтесь нашим графиком вакцинации, чтобы узнать, когда вашему ребенку будут делать каждую вакцинацию. Менингит — это инфекция, вызывающая отек головного и спинного мозга.
Другие состояния после рождения. Сюда входят:
- Травмы головы
- Желтуха, достаточно серьезная, чтобы потребовать переливания крови. Желтуха — это когда глаза и кожа ребенка желтеют. У ребенка желтуха, когда его печень не полностью развита или не работает. При переливании крови в ваше тело попадает новая кровь.
- Прием больших доз некоторых лекарств, например антибиотика стрептомицина. Антибиотики — это лекарства, убивающие определенные инфекции.
- Накопление ушной серы или жидкости за барабанной перепонкой
- Травма барабанной перепонки
- Предметы, застрявшие в слуховом проходе, например еда, игрушки или кусочки мелка
- Находясь рядом громкие звуки, похожие на звуки машин
Как лечить потерю слуха?
Лечение зависит от общего состояния здоровья вашего ребенка и причины потери слуха.Если у вашего ребенка потеря слуха, важно начать лечение как можно скорее, но не раньше, чем ему исполнится 6 месяцев. Это может помочь ей развить речь, язык и социальные навыки. Лечение может включать:
Кохлеарный имплант. Это небольшое электронное устройство, которое может помочь некоторым младенцам с тяжелой или глубокой потерей слуха. Одна часть имплантата находится на голове за ухом. Остальные части во время операции помещаются внутрь уха. Часть за ухом посылает звуки в части внутри уха.Младенцам в возрасте 1 года может быть установлен кохлеарный имплант. Это не дает ребенку полного слуха, но может дать ребенку ощущение звука. Слух через кохлеарный имплант отличается от обычного слуха. Такие специалисты, как аудиологи и логопеды, могут помочь вашему ребенку научиться слышать через имплант и развить его речь, язык и социальные навыки.
Ушные трубки. Это крошечные трубочки, проходящие через барабанную перепонку. Ушные трубки пропускают воздух в среднее ухо и предотвращают скопление жидкости за барабанной перепонкой.Вашему ребенку могут понадобиться ушные вкладыши, если:
- У нее много ушных инфекций (примерно три или более за 6 месяцев или четыре или более за 1 год)
- У нее скопление жидкости и воспаление (покраснение и припухлость) за барабанной перепонкой
- Если у нее потеря слуха из-за ушных инфекций
Слуховой аппарат. Слуховые аппараты делают звуки громче. Слуховые аппараты могут помочь детям в возрасте от 1 месяца. Если у вашего ребенка тяжелая или глубокая потеря слуха, слуховой аппарат может не помочь.
Изучение специальных языковых навыков. Детям с потерей слуха могут потребоваться специальные языковые навыки для общения с другими людьми. Например, некоторые могут выучить американский язык жестов (также называемый ASL) для общения. ASL использует форму, положение и движение рук, а также выражения лица и движения тела для общения.
Лекарственные средства, включая антибиотики. Если у вашего ребенка ушная инфекция, вызывающая потерю слуха, его лечащий врач может прописать антибиотик, например амоксициллин.Ее врач также может порекомендовать безрецептурные (также называемые безрецептурными) лекарства, которые помогут от лихорадки и боли. Эти лекарства могут включать ушные капли, ацетаминофен (торговая марка Tylenol®) и ибупрофен (торговые марки Motrin® или Advil®).
Логопед. Это терапия, чтобы научить вашего ребенка говорить более четко или общаться другими способами. Патологи речи (также называемые логопедами) — это профессионалы, которые могут помочь детям научиться издавать звуки, улучшить свой голос и взаимодействовать с другими.
Хирургия. Хирургия иногда может исправить проблемы со строением наружного и среднего уха.
Как предотвратить потерю слуха у ребенка?
Некоторые виды потери слуха, например, потерю слуха, вызванную изменениями генов, предотвратить невозможно. Но вы можете помочь предотвратить потерю слуха, связанную с другими причинами, такими как инфекции и преждевременные роды. Вот что вы можете сделать:
До беременности
- Пройдите предварительное обследование. Это медицинское обследование, которое вы должны пройти перед беременностью, чтобы убедиться, что вы здоровы, когда забеременеете.
- Убедитесь, что вы сделали своевременные прививки. Поговорите со своим врачом о вакцинации, которая вам необходима перед беременностью. Например, вам может потребоваться вакцинация MMR, которая защитит вас от кори и краснухи.
- Пройдите проверку на наличие инфекций, например ИППП. Раннее обследование и лечение могут помочь вам иметь здоровую беременность и родить здорового ребенка.
Во время беременности
Сходите на все медицинские осмотры во время беременности. Дородовая помощь — это медицинская помощь во время беременности. При каждом посещении дородовой помощи ваш лечащий врач проверяет вас и вашего растущего ребенка. Ранний и регулярный дородовой уход может помочь вам в благополучной беременности. Сходите на все медицинские осмотры, даже если вы чувствуете себя хорошо.
Защитите себя от инфекций. Вот что вы можете сделать:
- Если вам нужны прививки, сделайте их.Спросите своего врача о вакцинации, которую можно безопасно делать во время беременности.
- После посещения туалета или сморкания вымойте руки водой с мылом.
- Занимайтесь безопасным сексом. Это означает, что заниматься сексом только с одним человеком, у которого нет других половых партнеров. Если вы не уверены, есть ли у вашего партнера ИППП, используйте барьерный метод контроля над рождаемостью. Барьерные методы включают мужские и женские презервативы и зубные прокладки. Зубная прокладка — это квадратный кусок резины, который может помочь защитить вас от ИППП во время орального секса.
- Не ешьте недоваренное мясо.
- Не трогайте кошачьи корма.
После рождения
Возьмите вашего ребенка на все его медицинские осмотры. Во время этих осмотров лечащий врач проверяет общее состояние здоровья, рост и развитие ребенка. Вашему ребенку также делают прививки, чтобы защитить его от вредных инфекций.
Получите раннее лечение ушных инфекций. Если вы считаете, что у вашего ребенка ушная инфекция, немедленно позвоните его врачу.Признаки и симптомы ушной инфекции включают:
- Дергать за уши
- Проблемы со слухом или сном
- Плачет больше обычного, особенно когда ребенок лежит
- Проблемы с балансом
- Лихорадка
- Головная боль
- Жидкость выходит из ушей
Держите ребенка подальше от громких звуков. Закройте окна и двери в доме, чтобы не слышать громкие звуки рядом с ребенком. Снизьте громкость ТВ и радио.Подарите малышу тихие игрушки или игрушки с минимальной громкостью. Не водите малыша на шумные мероприятия, например на концерты или фейерверки. Если вы берете ребенка в шумное место, используйте беруши или наушники, чтобы уменьшить шум. Если ваш ребенок прикрывает уши или выглядит беспокойным в шумном месте, вероятно, лучше уйти.
Дополнительная информация
Последнее обновление: июнь 2019 г.
Унаследована ли глухота | Медицинский центр Калифорнийского университета в Ирвине
Глухота может быть наследственным заболеванием, которое присутствует при рождении ребенка.Унаследованная глухота также может развиваться с течением времени в детстве или во взрослом возрасте. По данным Американского фонда исследований слуха, около 1 из 1000–2000 рождений связано с врожденной глухотой. Рецессивное аутосомно-генетическое заболевание является причиной более 75% случаев врожденной глухоты. Существует множество различных генетических мутаций, которые могут вызвать глухоту, которая присутствует при рождении, и родители не всегда сразу понимают, что их дети страдают потерей слуха или глухими. Некоторые случаи генетической глухоты являются частью расстройства, которое вызывает другие нарушения, но примерно от 70 до 80% врожденной глухоты является отдельным заболеванием.
Синдромная наследственная глухота
По данным Центров по контролю за заболеваниями, от 20 до 30% людей, рожденных с глухотой, унаследованной от их родителей, также имеют другие виды инвалидности. Одна из наиболее распространенных инвалидностей, возникающих при глухоте, — это нарушения зрения. Младенцы, рожденные с глухотой, также могут иметь другие задержки в развитии, которые могут быть частью их генетической наследственности или из-за значительной недоношенности.
Когда ребенок рождается с генетическим заболеванием, вызывающим глухоту, важно, чтобы ребенок прошел тщательное обследование у врачей-сурдологов и сурдологов.Эти специалисты могут выявить сопутствующие заболевания, такие как нарушения речи и глотания, которые могут вызвать у ребенка дополнительные проблемы с ростом и развитием. Ранняя диагностика помогает ребенку получить раннее вмешательство. Услуги раннего вмешательства могут включать логопедию, слуховые аппараты, операцию по поводу кохлеарных имплантатов и другие услуги, которые помогут ребенку полностью раскрыть свой потенциал.
Как работает генетическое наследование глухоты
Аутосомно-рецессивная глухота в равной степени поражает мальчиков и девочек.Родитель, который может быть или не быть глухим, имеет 25% шанс передать мутацию ребенку. Есть несколько различных генетических механизмов, которые могут вызвать глухоту. Глухота, связанная с полом, возникает в результате мутации Х-хромосомы. Этот вид глухоты чаще всего выражается у мальчиков. Девочки страдают редко, потому что для глухоты у девочек, несущих две Х-хромосомы, требуются две копии мутировавшего гена. Американская академия отоларингологии объясняет, что аутосомно-доминантная потеря слуха встречается редко и поражает мальчиков и девочек в равном количестве.Родитель с геном имеет 50% шанс передать его своему ребенку.
Генетика несиндромальной врожденной потери слуха
Врожденное нарушение слуха поражает почти 1 из 1000 живорожденных и является наиболее частым врожденным дефектом в развитых странах. Наследственные типы потери слуха составляют более 50% всех случаев врожденной нейросенсорной тугоухости и вызваны генетическими мутациями. HL может быть несиндромным, ограничивающимся внутренним ухом, или синдромальным, как часть множества аномалий, влияющих на организм.Несиндромный HL можно разделить на категории по типу наследования, например, аутосомно-доминантный (называемый DFNA), аутосомно-рецессивный (DFNB), митохондриальный и X-связанный (DFN). На сегодняшний день в литературе описано 125 локусов глухоты: 58 локусов DFNA, 63 локуса DFNB и 4 X-сцепленных локуса. Мутации в генах, которые контролируют адгезию волосковых клеток, внутриклеточный транспорт, высвобождение нейромедиаторов, ионный гемеостаз и цитоскелет волосковых клеток, могут привести к нарушениям работы улитки и внутреннего уха. В последние годы с увеличением количества исследований генов, участвующих в врожденной потере слуха, появились и стали более доступными варианты генетического консультирования и лечения.В этой статье представлен обзор известных в настоящее время генов, связанных с несиндромной врожденной потерей слуха и мутациями во внутреннем ухе.
1. Введение
Потеря слуха (HL) — распространенное заболевание, и врожденное нарушение слуха поражает почти 1 из 1000 живорожденных; это одно из самых тяжелых расстройств и наиболее частый врожденный дефект в развитых странах [1]. Нарушение слуха влияет на развитие речи, овладение языком и образование у детей и, как следствие, часто приводит к уменьшению возможностей в трудовой жизни, поскольку люди с потерей слуха начинают изолироваться от общества.В США, по оценкам, социальные издержки нелеченой потери слуха в течение всей жизни могут достигать 1,1 миллиона на каждого человека, не получившего лечения [2]. Эти затраты могут быть уменьшены на 75 процентов при раннем вмешательстве и лечении [3].
Наследственная потеря слуха составляет почти 50% всех случаев врожденной нейросенсорной тугоухости и вызвана генетическими мутациями [4]. Глухота может быть результатом мутации одного гена или комбинации мутаций разных генов; это также может быть результатом причин окружающей среды, таких как травмы, лекарства, медицинские проблемы и воздействие окружающей среды, или результатом связи между факторами окружающей среды и генетикой [5].
HL может быть несиндромным, ограничивающимся внутренним ухом, или синдромным, частью множества аномалий, влияющих на организм. Несиндромный HL можно далее классифицировать по способу наследования. Примерно 20% несиндромной сенсоневральной тугоухости (NSSHL) наследуется по аутосомно-доминантному типу, также называемому DFNA; этот тип потери слуха обычно начинается отсроченно. Восемьдесят процентов наследственной HL является аутосомно-рецессивной (DFNB), при которой потеря слуха, как правило, является врожденной, но некоторые формы могут проявиться в более позднем возрасте.Наследование остальных типов HL является митохондриальным или X-сцепленным (DFN) (менее 1%) [2]. На сегодняшний день в литературе описано 125 локусов глухоты: 58 локусов DFNA, 63 локуса DFNB и 4 X-сцепленных локуса (http://hereditaryhearingloss.org/) [6].
Многие гены участвуют в функции внутреннего уха, и ухо очень чувствительно к мутациям в генетических локусах. Это связано с тем, что физиология и структура внутреннего уха уникальны и не похожи на другие анатомические области. Мутации в генах, которые контролируют адгезию волосковых клеток, внутриклеточный транспорт, высвобождение нейромедиаторов, ионный гемеостаз и цитоскелеты волосковых клеток, могут привести к нарушениям работы улитки и внутреннего уха.
В последние годы с увеличением количества исследований генов, участвующих в врожденной потере слуха, появились и стали более доступными варианты генетического консультирования и лечения. В диагностических тестах часто участвуют гены, которые являются частыми причинами потери слуха, такие как GJB2 , GJB6 , SLC26A4 и OTOF [7]. Результаты этих тестов можно использовать при консультировании родителей относительно прогноза потери слуха у ребенка, прогнозировании рецидива у будущего потомства и принятии во внимание терапевтических возможностей, таких как кохлеарная имплантация [2].В недавних исследованиях некоторые вирусные векторы были доставлены во внутреннее ухо, чтобы заменить нормальную копию гена дефектным геном, вызывающим потерю слуха. В исследовании на животных было обнаружено, что доставленный аденовирусом SLC17A8 (VGLUT-3; везикулярный транспортер глутамата 3) восстанавливает слух у мышей. В другом исследовании развитие и регенерация волосковых клеток были вызваны доставкой гена ATOh2 [8, 9].
В этом мини-обзоре представлен обзор и описаны известные в настоящее время гены, связанные с несиндромной врожденной потерей слуха и мутациями, вызывающими нарушение функции белков во внутреннем ухе (Таблица 1).
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DFNA = несиндромальная доминантность ; DFNB = несиндромная глухота, аутосомно-рецессивная; AD = аутосомно-доминантный; AR = аутосомно-рецессивный; TM = текториальная мембрана; ЕСМ = внеклеточный матрикс; Ca +2 = ион кальция; K + = ион калия. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
2.Гены и белки, связанные с несиндромной потерей слуха
2.1. Белки адгезии
Стереоцилии волосковых клеток в улитке связаны и связаны с текториальной мембраной с помощью различных белков адгезии. Пучки волос стабилизируются набором временных звеньев, таких как переходные боковые звенья и звенья голеностопного сустава. Эти связи также вызывают рост и созревание сигнальных комплексов [10]. В зрелых волосковых клетках стереоцилии соединяются с помощью текториальных прикрепляющих коронок, горизонтальных верхних соединителей и концевых звеньев [2].На сегодняшний день сообщается о нескольких генах, относящихся к связывающему аппарату. Это DFNA4 ( CEACAM16 (молекула канцерогенной антиген-связанной клеточной адгезии 16)) [11], DFNB12 ( CDh33 (кадгерин 23)) [12], DFNB16 ( STRC (стереоцилин)) [1318], DFNB ( USh2C (гармонин)) [14, 15], DFNB22 ( OTOA (отоанкорин)) [16], DFNB23 ( PCDh25 (протокадгерин 15)) [17], DFNB31 ( WHRN ) (whirlin ) [18], DFNB66 / 67 ( TMHS, (тетраспановый мембранный белок)) [19] и DFNB84 ( PTPRQ (рецептор Q тирозинфосфата)) [20].
Гены PTPRQ и TMHS , а также кадгерин 23 и протокадгерин 15 являются частями временного латерального звена. Во время развития они препятствуют слиянию каждой стереоцилии сами по себе [2]. В зрелых волосковых клетках они становятся основными частями концевого звена и действуют как ворота, управляя механотрансдукцией и обеспечивая стабильность, играя центральную роль в слуховой функции [21].
Whirlin и гармонин регулируют связующие комплексы и служат каркасными белками.Мутации в этих белках вызывают потерю слуха по аутосомно-рецессивному типу, но Sans, который является третьим каркасным белком, связан со сложной синдромальной потерей слуха, синдромом Ашера. Другие гены, USh3 α и VLGR1b , также связаны с синдромом Usher, и они являются частью стереоцилиального звена голеностопного сустава [22].
Стереоцилин — это белок внеклеточного матрикса, который прикрепляет самые высокие стереоцилии наружных волосковых клеток к текториальной мембране [13].Место прикрепления этой текториальной мембраны обычно образовано CEACAM16 . Подобным образом отоанкрин также прикрепляет несенсорные клетки к текториальной мембране [16].
2.2. Транспортные белки
Во внутреннем ухе все части семейства миозинов могут использоваться для транспортировки различных белков. При использовании АТФ эти миозиновые белки связываются с актиновым цитокелетом и продвигаются вперед. Сайты связывания переносимых белков находятся на карбоксильных концах транспортных белков [23].К миозинам, связанным с наследственной потерей слуха, относятся миозин Ia (DFNA48) [24], миозин IIIa (DFNB30) [25], миозин VI (DFNA22 / DFNB37) [26, 27], миозин VIIa (DFNA11 / DFNB2) [28, 29 ], немышечной тяжелой цепи IX миозина (DFNA17) [30], немышечной тяжелой цепи XIV миозина (DFNA4) [31] и миозина XVa (DFNB3) [32]. Все они имеют свои уникальные функции в волосковых клетках внутреннего уха [2].
2.3. Белки синапсов
VGLUT3, который является везикулярным рецептором глутамата, играет роль в синапсах внутренних волосковых клеток.Он кодируется SLC17A8 в локусе DFNA25 и связан с аутосомно-рецессивной потерей слуха [33]. Этот белок регулирует как экзоцитоз, так и эндоцитоз глутамата. Отоферлин (кодируемый OTOF ) представляет собой белок, который работает с миозином VI в синаптической щели внутренней волосковой клетки и играет роль в кальций-зависимом слиянии везикул с плазматической мембраной. В результате высвобождается глутамат и возбуждается афферентный нейрон [34]. В исследовании на животных с участием мышей с нокаутом OTOF и SLC17A8 наблюдалось снижение количества постсинаптических ганглиозных клеток, и был сделан вывод, что эти белки очень важны для сохранения и развития нормального слуха [35].
2.4. Электромобильность
Улитка чувствительна и избирательна к звукам, издаваемым внешними волосковыми клетками. Это происходит с помощью процесса, называемого электродвижением, и считается, что за это отвечает белок под названием престин [2]. Он изменяет потенциал мембраны и позволяет изменять длину наружных волосковых клеток. Когда это происходит, внешняя волосковая клетка становится длиннее при гиперполяризации и короче при деполяризации, поэтому она усиливает свою чувствительность к звуку [36].Этот белок кодируется как SLC26A5 и впервые был описан Zheng et al. в 2000 г. [37]. Мутации в SLC26A5 являются причиной потери слуха DFNB61 [38].
2.5. Cytoskeleton
Мутации в некоторых генах, связанных с организацией цитоскелета, могут вызывать NSSHL; это ESPN (эспин), RDX (радиксин), TRIOBP (триосвязывающий белок), ACTG1 ( γ -актин), TPRN (таперин), DIAPH2 и SMPX (белок малых мышц, Х-связанный).
Белок espin обеспечивает стабильность стереоцитоскелета. Мутация ESPN может вызывать DFNB36 и аутосомно-доминантную тугоухость [39]. С помощью радиксина можно добиться большей стабильности стериоциллий. Он связывает актиновые филаменты с плазматической мембраной и располагается вдоль стереоцилий. Мутации в RDX могут вызывать DFNB24 и аутосомно-рецессивную глухоту [40]. γ -актин действует как строительный блок для стереоцилий волосковых клеток. Эти стереоцилии постоянно подвергаются деполимеризации у основания и полимеризации актина на кончике [41].Мутации в ACTG1 могут вызывать DFNA20 / 26 и аутосомно-доминантную тугоухость [42, 43]. Благодаря постоянному процессу ремоделирования другие белки также важны для непрерывности. Diaphanous 1 регулирует реорганизацию и полимеризацию мономеров актина в полимеры. Он кодируется как DIAPh2 , и мутации в этом гене могут вызывать DFNA1 и аутосомно-доминантную тугоухость [44]. Связывание и организация γ -актина в основании стереоцилий обеспечивается двумя изоформами гена TRIOBP .Мутации в изоформах TRIOBP4 и TRIOBP5 могут вызывать DFNB28 и тугоухость аутосомно-рецессивного типа [45, 46]. Другой белок, таперин, локализован в основании стереоцилий и ассоциирован с DFNB79 [47]. Х-связанный белок малых мышц, кодируемый как SMPX (DFN4), выполняет функцию стереоцилий в развитии и поддержании в ответ на механический стресс, которому подвергаются стереоцилии [48].
2.6. Ионный гомеостаз и щелевые соединения
В улитке есть два типа жидкостей: перилимфа с высоким содержанием натрия и низким содержанием калия и эндолимфа с высоким содержанием калия и низким содержанием натрия; это состояние создает очень положительный потенциал (+80 мВ), называемый эндокохлеарным потенциалом.Приток калия в волосковые клетки вызывает деполяризацию, после чего волосковая клетка реполяризуется и перемещает катионы обратно в эндолимфу. Этот ионный гомеостаз включает белок плотных контактов 2 ( TJP2 ), трицеллулин ( MARVELD2 / TRIC ), клаудин 14 (CLDN14), KCNQ4 ( KCNQ4 ), Барттин ( BSND41842b2), ATP / PMCA2 ), некоторые коннексины ( GJBs ) и пендрин ( SLC26A4 ), и все они связаны с наследственной потерей слуха [2].
В мутации CLDN14 в DFNB29 белок claudin 14 будет отсутствовать или дисфункциональным, а пространство Nuel, которое окружает базолатеральную поверхность наружных волосковых клеток, затронуто и может изменить его электрический потенциал [49]. Точно так же трицеллулин, который кодируется как MARVELD2 / TRIC , вызывает DFNB49 при мутации, и он функционирует как плотное соединение, которое соединяет клетки вместе [45]. Белок плотных соединений 2, кодируемый как ген TJP2 , связывает плотные соединения с актиновым цитоскелетом, а мутации вызывают DFNA51 и тугоухость аутосомно-доминантного типа [50].
KCNQ4 кодирует белок, образующий потенциал-зависимый калиевый канал. Он экспрессируется в наружных волосковых клетках и в случае мутации вызывает HL аутосомно-доминантного типа, DFNA2a [51]. Он помогает в реполяризации наружных волосковых клеток и регулирует чувствительность к звуку.
Барттин и пендрин, кодируемые как BSND и SLC26A4 , соответственно, участвуют как в несиндромном, так и в синдромальном HL. Пендрин является анионообменником и играет решающую роль в кислотно-щелочном балансе.И синдромный (синдром Пендреда, связанный с зобом), и несиндромный HL (DFNB4) связаны со степенью мутации в SLC26A4 [52]. Белок Барттин — одна из субъединиц хлоридного канала. В основном, мутации в BSND могут вызывать синдром Барттера, связанный с потерей слуха и почечными аномалиями, но DFNB73 также приписывается мутации в BSND и вызывает несиндромную глухоту [53].
Щелевой переход — это канал, проходящий через две соседние мембраны, который обеспечивает обмен различными молекулами и ионами в улитке.Эти соединения состоят из белков, называемых коннексинами. Эти соединения также играют роль в рециркуляции ионов калия, необходимых для нормального слуха. это наиболее частая причина несиндромного HL, и первым идентифицированным геном является GJB2; он кодируется как коннексин 26 (DFNA3a / DFNB1a) [54]. Другими коннексинами, относящимися к несиндромному HL, являются коннексин 30 ( GJB6 , DFNA3b / DFNB1b) [55, 56] и коннексин 31 ( GJB3 , DFNA2b / DFNB91) [57, 58].
2.7. Другие
Существуют также белки внеклеточного матрикса, то есть TECTA ( α -текторин), COL11A2 (коллаген XI типа α 2) и COCH (кохлин), и факторы транскрипции, такие как как POU4f3 (класс 4 POU), POU3f4 (класс 3 POU), MIR96 (микроРНК 96), GRHL2 (зернистая головка 2), ESRRB (рецептор, связанный с эстрогеном 42 β 908 ), и EYA4 (глаза отсутствуют 4), вовлеченные в наследственный HL.
3. Заключение
В этом обзоре представлен обзор и описание известных в настоящее время генов, относящихся к наследственному NSSHL. Со временем функции этих генов будут лучше поняты, и вскоре будет обнаружено больше генов, ведущих к потере слуха. С новыми исследованиями и постоянным обследованием функция улитки будет лучше изучена, и, как мы надеемся, будут разработаны новые молекулярные и генные методы лечения нейросенсорной HL человека.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов в отношении публикации данной статьи.
Глухота и потеря слуха
Более 5% населения мира — или 430 миллионов человек — нуждаются в реабилитации для решения проблемы их «инвалидизирующей» потери слуха (432 миллиона взрослых и 34 миллиона детей). По оценкам, к 2050 году более 700 миллионов человек — или каждый десятый человек — страдает потерей слуха.
«Отключение» потери слуха означает потерю слуха более 35 децибел (дБ) в ухе с лучшим слухом. Почти 80% людей с потерей слуха живут в странах с низким и средним уровнем дохода.Распространенность потери слуха увеличивается с возраст, среди лиц старше 60 лет более 25% страдают потерей слуха.
Потеря слуха и глухота
Считается, что человек, который не может слышать так же хорошо, как человек с нормальным слухом (порог слышимости на оба уха составляет 20 дБ или выше), страдает потерей слуха. Потеря слуха может быть легкой, средней, тяжелой или глубокой. Это может повлиять на одно ухо или оба уха и приводит к затруднению слышимости разговорной речи или громких звуков.
Термин «слабослышащий» относится к людям с потерей слуха от легкой до тяжелой. Люди с нарушениями слуха обычно общаются с помощью разговорной речи и могут воспользоваться слуховыми аппаратами, кохлеарными имплантатами и другими вспомогательными устройствами, а также субтитрами.
«Глухие» люди в основном страдают глубокой потерей слуха, что означает очень слабый слух или его отсутствие. Они часто используют язык жестов для общения.
Причины потери слуха и глухоты
Хотя эти факторы могут встречаться в разные периоды жизни, люди наиболее восприимчивы к их воздействию в критические периоды жизни.
Пренатальный период
- Генетические факторы — включая наследственную и ненаследственную тугоухость
- Внутриутробные инфекции — такие как краснуха и цитомегаловирусная инфекция
Перинатальный период
- Асфиксия при рождении время рождения
- Гипербилирубинемия (тяжелая желтуха в неонатальном периоде)
- Низкая масса тела при рождении
- Другие перинатальные заболевания и их лечение
Детский и подростковый возраст
- Хронические инфекции уха (хронические гнойные отиты) media)
- Сбор жидкости в ухе (хронический негнойный средний отит)
- Менингит и другие инфекции
Взрослый и пожилой возраст
- Хронические заболевания
- Курение
- Отосклероз 9005 5
- Возрастная сенсоневральная дегенерация
- Внезапная сенсоневральная потеря слуха
Факторы на протяжении всей жизни
- Церезумия (поражение ушной серы)
- Травма уха или головы
- Громкий шум / громкие звуки
- Ототоксические лекарства
- Ототоксические химические вещества, связанные с работой
- Недостаток питательных веществ
- Вирусные инфекции и другие заболевания уха
- Отсроченное начало или прогрессирующая генетическая потеря слуха
Влияние неуправляемой потери слуха
Без внимания потеря слуха влияет на многие аспекты жизни на индивидуальном уровне :
Коммуникация и речь
Познание
Образование и работа: В развивающихся странах дети с потерей слуха и глухотой часто делают это. не г получить образование.Взрослые с потерей слуха также имеют гораздо более высокий уровень безработицы. Среди тех, кто работает, более высокая процент людей с потерей слуха находится на более низких ступенях занятости по сравнению с общей рабочей силой.
Социальная изоляция, одиночество и стигма
Влияние на общество и экономику
Количество лет, прожитых с инвалидностью (YDL), и лет жизни с поправкой на инвалидность (DALY)
По оценкам ВОЗ, неизлеченная потеря слуха влечет за собой ежегодные глобальные затраты в США. 980 миллиардов долларов.Сюда входят расходы сектора здравоохранения (за исключением стоимости слуховых аппаратов), расходы на поддержку образования, снижение производительности и социальные издержки. 57% из них затраты относятся на страны с низким и средним уровнем доходов.
Профилактика
Многих причин, приводящих к потере слуха, можно избежать с помощью стратегий общественного здравоохранения и клинических мероприятий, проводимых на протяжении всей жизни.
Профилактика потери слуха необходима на протяжении всей жизни — от пренатального и перинатального периодов до пожилого возраста.У детей почти 60% потери слуха вызваны причинами, которых можно избежать, которые можно предотвратить с помощью мер общественного здравоохранения.
меры. Аналогичным образом, у взрослых можно предотвратить наиболее частые причины потери слуха, такие как воздействие громких звуков и ототоксичных лекарств.
К эффективным стратегиям снижения потери слуха на разных этапах жизни относятся:
- иммунизация;
- надлежащая практика охраны здоровья матери и ребенка;
- генетическое консультирование;
- идентификация и лечение общих заболеваний уха;
- профессиональные программы сохранения слуха от шума и химического воздействия;
- стратегии безопасного прослушивания для уменьшения воздействия громких звуков в условиях отдыха; и
- рациональное использование лекарственных средств для профилактики ототоксической потери слуха.
Выявление и лечение
- Раннее выявление потери слуха и заболеваний уха является ключом к эффективному лечению.
- Это требует систематического обследования для выявления потери слуха и связанных с ней заболеваний уха у тех, кто подвергается наибольшему риску. Это включает:
- Новорожденные и младенцы
- Дети дошкольного и школьного возраста
- Люди, подвергающиеся воздействию шума или химических веществ на работе
- Люди, получающие ототоксические лекарства
- Пожилые люди
- Оценка слуха и проверка уха могут проводиться в клинических и настройки сообщества.Такие инструменты, как приложение ВОЗ «HearWHO» и другие технологические решения, позволяют проводить скрининг заболеваний уха и потери слуха с помощью ограниченное обучение и ресурсы.
- После выявления потери слуха важно устранить ее как можно раньше и надлежащим образом, чтобы смягчить любое неблагоприятное воздействие.
- Меры, доступные для реабилитации людей с потерей слуха, включают:
- использование слуховых технологий, таких как слуховые аппараты, кохлеарные имплантаты и имплантаты среднего уха;
- использование языка жестов и других средств сенсорной замены, , таких как чтение речи, использование печати на ладони или тадома, подписанное общение; и
- реабилитационная терапия для улучшения навыков восприятия и развития коммуникативных и языковых способностей.
- Использование вспомогательных слуховых аппаратов и услуг, таких как системы частотной модуляции и петли, устройства оповещения, телекоммуникационные устройства, услуги субтитров и перевод жестового языка, может еще больше улучшить доступ к общению и образованию для людей с потерей слуха.
Ответ ВОЗ
Работа ВОЗ в области ухода за ухом и слухом направлена на продвижение интегрированной помощи уха и слуха, ориентированной на людей (IPC-EHHC).
В своей работе ВОЗ руководствуется рекомендациями Всемирного доклада ВОЗ о слухе (2021 г.) и резолюцией Всемирной ассамблеи здравоохранения о профилактике глухоты и потери слуха.
Работа ВОЗ включает:
- направлять, помогать и поддерживать государства-члены в повышении осведомленности по вопросам EHC;
- способствовать созданию и распространению данных и информации, касающихся ухода за ухом и слухом;
- предоставляет технические ресурсы и рекомендации для содействия планированию и наращиванию потенциала систем здравоохранения в области ухода за слухом и слухом;
- поддержка обучения медицинского персонала уходу за слухом и слухом;
- содействие безопасному прослушиванию для снижения риска потери слуха, вызванной шумом в развлекательных целях, в рамках инициативы ВОЗ «Сделать прослушивание безопасным»;
- проведение Всемирного дня слуха как ежегодного информационно-пропагандистского мероприятия;
- налаживание партнерских отношений для разработки эффективных программ по уходу за слухом, включая инициативы по доступным слуховым аппаратам, кохлеарным имплантам и услугам;
- Защита ушей и слуха на Всемирном форуме по слухам.
Границы | Врожденная глухота приводит к изменению явного глазодвигательного поведения
Введение
Хорошо известно, что окуломоторное поведение зависит как от зрительной, так и от вестибулярной информации, но есть также некоторые свидетельства того, что слуховые функции участвуют в окуломоторном поведении (например, Paulsen and Ewertsen, 1966; Rolfs et al., 2005; Valsecchi and Turatto, 2009; Kerzel et al., 2010; Yuval-Greenberg, Deouell, 2011; Zou et al., 2012). Например, хорошо известно, что вращение звука вокруг головы субъекта может вызвать нистагм (Paulsen and Ewertsen, 1966). Несколько исследований показали, что слуховой стимул может приводить к визуальным саккадам в направлении источника звука (Zahn et al., 1978; Zambarbieri et al., 1982; Van Grootel and Van Opstal, 2009) и что предъявление слухового стимула может снижают частоту саккад (Rolfs et al., 2005; Kerzel et al., 2010; Yuval-Greenberg and Deouell, 2011; Zou et al., 2012).Более того, одно исследование показало доказательства того, что положение взгляда может влиять на точность слуховой локализации (Maddox et al., 2014), а результаты нашей команды показывают, что слушание или даже воображение звукового стимула движения может вызывать непроизвольные движения глаз (Landry et al., 2015) . Учитывая очевидную связь между слуховым входом и окуломоторным поведением, можно задаться вопросом, как потеря слухового сигнала от рождения может повлиять на это моторное поведение.
Есть два способа изучения глазодвигательного поведения.Как описано Munoz и Coe (2011), явное, глазодвигательное поведение просто состоит из движения глаз. Напротив, окуломоторное поведение скрытого визуального отбора более сложное. Они подразумевают переключение внимания с движением глаз или без него. Во всех экспериментах по изучению глазодвигательного поведения глухих использовались парадигмы, изучающие аспекты визуального внимания движений глаз (например, Heimler et al., 2015; Prasad et al., 2015; Jayaraman et al., 2016). Большинство этих исследований демонстрируют повышенное избирательное внимание к цели на периферии зрения или к движущейся цели у глухих.
Отслеживание движения глаз позволяет выделить несколько компонентов, включая саккады и плавное преследование (Leigh and Zee, 2015). Саккады и плавное преследование различны и отражают независимые процессы (саккады: Sparks and Mays, 1990; преследование: Lisberger et al., 1987). Только в одном исследовании измерялась работа глаз у глухих с помощью классического открытого задания на глазодвигательное поведение — про- и антисаккад (Bottari et al., 2012). Результаты показали более быструю латентность саккад и меньшую частоту ошибок в исследованиях против саккад у глухих, что указывает на возможное изменение баланса между произвольным и рефлексивным движением глаз.Открытое глазодвигательное поведение у глухих в дальнейшем не исследовалось. В отличие от саккад, плавное преследование требует постоянного регулирования с помощью петель обратной связи (Leigh and Zee, 2015). Плавное преследование позволяет увидеть такие процессы, как создание движений, целостность комбинированных контуров визуальной и моторной обратной связи и нарушение контроля с помощью обратной связи (например, Robert et al., 2014; Lizak et al., 2016). Здесь мы стремились изучить точность плавного преследования и саккады у глухих.
Методы
Участников
В настоящем исследовании приняли участие 24 взрослых.Двенадцать из них были глухими и страдали от тяжелой или глубокой потери слуха. Девять из этих испытуемых были глухими от рождения. Три других субъекта стали глухими в младенчестве, от 2 до 24 месяцев (один от неизвестного заболевания, один от наследственного состояния и один от преждевременных родов) (возрастной диапазон 18–42, M = 29). У всех участников была длительная тяжелая и глубокая сенсоневральная потеря слуха. Когда они пришли на исследование, у них была с собой последняя аудиограмма. Эти пороги использовались. Все аудиограммы были сделаны в течение года до исследования.Все участники, кроме одного, использовали двусторонний слуховой аппарат и использовали устную речь в дополнение к чтению по губам. Единственным участником, который использовал язык жестов, был тот, кто не использовал слуховой аппарат (характеристики участников см. В Таблице 1). Двенадцать контрольных субъектов имели нормальный слух и не имели отологических проблем (возрастной диапазон 18–34 лет, M = 26). У всех субъектов было нормальное зрение или зрение с поправкой на нормальное. Зрение проверялось с помощью глазной диаграммы исследования раннего лечения диабетической ретинопатии (ETDRS) на расстоянии 1.5 метров. Установленный критерий составлял 20/20 для каждого глаза либо для нормального, либо для скорректированного до нормального зрения. Все участники получили высшее образование. Ни у кого из участников из обеих групп не было нарушений обучаемости, неврологических проблем или других известных заболеваний. Все участники были добровольными добровольцами и лечились в соответствии с Заявлением о политике Tri-Council: этическое поведение в исследованиях с участием людей (Совет медицинских исследований Канады MRRC, 2003). Все участники были наивны по отношению к цели эксперимента.
Таблица 1. Характеристики участников.
Записи движения глаз
Положение глаза было получено неинвазивным способом с использованием системы EyeLink 1000 на основе видео с камерой с обновленной частотой 2000 Гц (SR Research, Канада). Система EyeLink 1000 регистрирует положение бинокулярного глаза с частотой дискретизации 1000 Гц и пространственным разрешением <0,01 °. Движения головы ограничивались использованием упора для подбородка, расположенного на расстоянии 60 см от линеаризованного видеомонитора (Viewsonic 19 ″ CRT, разрешение 1024 × 768 пикселей, частота обновления 100 Гц).Процедура калибровки по девяти точкам выполнялась в начале каждого экспериментального условия с использованием местоположений EyeLink по умолчанию. Калибровку повторяли, если какая-либо точка калибровки отклонялась от целевого значения более чем на 1 ° или если средняя ошибка для всех точек превышала 0,5 °. Средняя точность калибровки по всем участникам составила 24 мин. дуги для центральной калибровочной точки и 32 мин. дуги крайних точек калибровки по углам экрана.
Перед началом эксперимента экспериментатор устно излагал соответствующие инструкции и выводил их на экран, чтобы обеспечить полное понимание экспериментальной задачи всеми участниками.
Участники смогли четко понимать устные инструкции, используя сочетание чтения по губам и слуховых аппаратов. Для одного участника, который общается только с помощью языка жестов, инструкции были четко написаны. Инструкции для каждого условия были представлены в письменной форме на экране компьютера, что позволило всем участникам их прочитать. У них было столько времени, сколько им было необходимо, чтобы прочитать и понять инструкции, и экспериментатор удостоверился, что инструкции были поняты до начала эксперимента.Ни один из слабослышащих участников не сообщил о каких-либо существенных затруднениях при чтении.
Эксперимент: задача преследования
Участников попросили отслеживать движущуюся круговую цель 0,5 °. Коррекция дрейфа была достигнута за счет смещения цели на 10 °, при этом местоположение зависело от типа движения.
Цель коррекции сноса началась в другом месте в зависимости от типа движения. Например, для цели, движущейся вверх / вниз, влево / вправо, начальная коррекция сноса была в центре экрана.При движении по часовой стрелке / против часовой стрелки цель появлялась в левой части эллипса.
После коррекции смещения и пустого экрана 50 мс участникам было предложено двигать глазами, чтобы следить за целью, когда она двигалась по экрану в одном из четырех направлений: горизонтально, вертикально, эллиптически по часовой стрелке или эллиптически против часовой стрелки. Более того, цель двигалась с одной из двух скоростей (2 или 4 град / с), в результате чего восемь условий повторялись трижды случайным образом.Интервал между испытаниями задавался самостоятельно, так как участник должен был смотреть на маркер фиксации и нажимать клавишу пробела, чтобы начать следующее испытание. Пример исходных данных для обеих групп см. На Рисунке 1.
Рисунок 1. Примеры необработанных пространственных данных о движениях глаз для одного контрольного участника (A) и одного участника с нарушением слуха (B) . (C) — временной ряд, представляющий один цикл целевого эллиптического движения в положениях X (черный) и Y (синий) на экране, и соответствующие движения глаз отслеживания в координатах X (красный) и Y (зеленый). (D) То же, что и C, но для слабослышащих участников. Отсутствующие данные отражают недостающие образцы из-за моргания глаз. Обратите внимание, что все четыре панели представляют экспериментальные условия, при которых стимул двигался против часовой стрелки с низкой скоростью.
Положение цели менялось в зависимости от задачи. При перемещении вверх / вниз, влево / вправо цель начиналась в центре экрана (512 384 пикселя в координатах X / Y). В состоянии вверх / вниз цель затем переместится на 5 ° вверх, остановится, затем на 10 ° вниз, остановится и, наконец, на 5 ° вверх, чтобы снова остановиться в центре экрана.То же верно и для левого / правого состояния.
При эллиптическом движении цель двигалась в том же направлении (по или против часовой стрелки), образуя круг с общей длиной радиуса 20 °. Во всех случаях движущийся стимул предъявлялся на 10 ° с. Для обоих эллиптических условий цель стартовала в координатах в левой части экрана (171 384), при этом цель продолжала вращаться либо по часовой стрелке, либо против часовой стрелки из этой позиции. Параметры эллипса: Амплитуда оси X 341 пиксель.Амплитуда оси Y 256 пикселей. Мишень была белой (RGB = 255,255,255), фон был черным (RGB = 0,0,0).
Анализ данных
Использовались данные только для правого глаза. Периоды мигания определялись с помощью алгоритма эвристической фильтрации EyeLink 1000 (Stampe, 1993) и удалялись. Кроме того, все образцы за 200 мс до и после каждого моргания были удалены, чтобы исключить начальную и конечную фазы моргания, во время которых зрачок мог быть частично закрыт. Мы также удалили части данных, которые содержали очень быстрое увеличение и уменьшение площади зрачка (20 единиц на выборку).Мартинес-Конде и др. (2006) идентифицировали эти периоды как частичные мигания, которые не полностью закрывают зрачок и которые не обнаруживаются алгоритмом Eyelink 1000. Остальные данные о движении глаз были проанализированы после очистки данных моргания и частичного моргания.
Для задачи преследования точность измерялась путем расчета способности человека сохранять фиксацию в пределах радиуса 1 ° от цели во время преследования. Это вычислялось каждые 20 образцов (т.е. каждые 20 мс), и для каждого испытания была получена процентная точность.Анализ проводился с использованием динамической интересующей области, которая отслеживала положение цели на каждой выборке айтрекера. Люди были классифицированы как точные, если их положение глаз находилось в пределах окна 1 ° вокруг цели в течение периода выборки, или неточные, если их положение глаз превышало окно 1 °.
Затем точность (и все последующие зависимые переменные) были введены в линейную модель смешанных эффектов. Модели с линейными смешанными эффектами могут учитывать вариации между участниками и между элементами, включая случайные эффекты в дизайн модели, которые вносятся вариацией из-за индивидуальных различий.Их принципиальные методы моделирования гетероскедастичности и несферической дисперсии ошибок придают линейным моделям смешанных эффектов большую мощность, чем традиционные меры (Baayen et al., 2008). Были построены отдельные линейные модели смешанного эффекта по точности для каждого из четырех направлений (горизонтальное, вертикальное, эллиптическое по часовой стрелке и эллиптическое против часовой стрелки). Чтобы предсказать точность, мы включили один фактор между субъектами с двумя уровнями (слабослышащий и контрольный) и фактор скорости внутри субъектов с двумя уровнями (2 и 4 град / с) в качестве фиксированных и случайных эффектов, что позволило им варьируются от участников.Обратите внимание, что мы использовали метод Саттервейта для определения степеней свободы. Помимо точности, мы также рассчитали модели смешанного эффекта со следующими зависимыми переменными (1) количество саккад за испытание, (2) латентность саккады (время от начала стимула до начала первой саккады), (3) саккада амплитуда, (4) продолжительность саккады и (5) средняя скорость саккады для каждого из четырех направлений. В дополнение к моделям смешанного эффекта, анализ post hoc включает Hedges ’ g в качестве меры величины эффекта, подходящей для небольших размеров выборки, и двусторонние тесты Манна-Уитни.
Мы использовали линейную модель, потому что при больших размерах выборки бином хорошо аппроксимируется распределением Гаусса. Чтобы подтвердить это, мы оценили нормальность этих данных с помощью критерия Шаприо-Уилка (все p > 0,05, указывающие на то, что эти данные существенно не отличались от нормального распределения). Кроме того, линейность распределения на графике квантиль – квантиль (Q – Q) подтвердила, что эти данные имеют нормальное распределение. После удаления миганий в среднем 93% информации об образце было сохранено для использования в анализе.
Результаты
На рисунке 2 представлена точность, измеренная в условиях преследования для группы с нарушениями слуха (светлые столбцы) и группы с нормальным контролем слуха (темные столбцы) для каждой из четырех задач преследования (вертикальная, горизонтальная, эллиптическая по часовой стрелке и эллиптическая. против часовой стрелки) с двумя скоростями: медленной (2 ° с –1 ) и быстрой (4 ° с –1 ). Точность отслеживания цели измерялась как процент от 10-секундного испытания, когда участник сохранял фиксацию в пределах радиуса 1 ° от цели.Анализ точности выполнения задания показывает существенные различия между группами, F (1,33) = 11,082, p = 0,002, а между скоростями F (1,31,2) = 9,507, p = 0,004, но без взаимодействия, F (1,33) = 0,888, p = 0,353, во время задачи эллипса по часовой стрелке. Точно так же был главный эффект группы, F (1,33) = 12,34, p = 0,001, и скорость, F (1,30,7) = 15,28, p <0.001, во время задания эллипса против часовой стрелки, но без взаимодействия, F (1,33) = 2,61, p = 0,116. Как видно на рисунке 2, не было никакого эффекта группы, никакого эффекта скорости и никакого взаимодействия во время горизонтального и вертикального преследования (все p > 0,1). Апостериорные сравнения показывают уменьшение средней разницы в точности между контрольной группой и участниками с нарушением слуха для медленного эллипса по часовой стрелке ( M diff = -15,440 [95% ДИ -25,544, -5.976], Манн Уитни U = 118, p = 0,009 , г Хеджеса = -1,175 [95% ДИ -1,928, -0,256]) и медленный эллипс против часовой стрелки ( M diff = -17,382 [95% ДИ -26,884, -8,3], Манна Уитни U = 122, p = 0,004, г Хеджеса = -1,375 [95% ДИ -2,137, -0,502]).
Рис. 2. Результаты точности для условий преследования для группы с нарушениями слуха (светло-серый) и контрольной группы (темно-серый).Средняя разница в точности (% времени достижения цели) для всех восьми условий показана на приведенном выше графике оценки Камминга. Необработанные данные нанесены на верхние оси; каждое среднее различие нанесено на нижние оси как распределение выборки начальной загрузки (было взято 5000 выборок начальной загрузки; доверительный интервал скорректирован на систематическую ошибку и ускорен). Средние различия обозначены точками; 95% доверительные интервалы обозначены концами вертикальных полос погрешностей.
Мы также проанализировали различные измерения, которые влияют на движение глаз во время преследования, в частности количество, продолжительность, задержку, амплитуду и скорость саккад.Для среднего количества саккад (см. Рисунок 3), выполненных в задаче с эллипсом против часовой стрелки, был основной эффект группы F (1,32,7) = 6,047, p = 0,019 и скорости F ( 1,17) = 37,284, p <0,001, но нет взаимодействия, F (1,33) = 0,505, p = 0,482 . Также был основной эффект скорости в состоянии по часовой стрелке F (1,32,1) = 65,77, p <0,001, и основной эффект группы F (1,28.3) = 8,83, p = 0,006. Интересно (с учетом результатов точности задания) было главное влияние скорости, F (1,19,2) = 80,662, p <0,001, для задачи вертикального преследования, но не было главного эффекта группы, F (1,11,2) = 1,694, p = 0,219. В горизонтальном положении были значительные основные эффекты скорости, F (1,13,7) = 62,02, p ≤ 0,001, но не группа, F (1,5,2) = 2,80, p = 0.115. Кроме того, анализ не выявил какого-либо значимого взаимодействия (все p > 0,1) между группой и скоростью в любой из четырех задач преследования. Для всех других показателей движения глаз (среднее значение амплитуды саккад, время ожидания до первой саккады, продолжительность и скорость саккад) модели смешанного эффекта не показали основного эффекта группы для всех условий (вертикального, горизонтального, эллиптического по часовой стрелке и эллиптический против часовой стрелки: p > 0,1).
Рисунок 3. Результаты Saccade для условий преследования для группы с нарушениями слуха (светло-серый) и контрольной группы (темно-серый). Средняя разница количества саккад для всех восьми условий показана на приведенном выше графике оценки Камминга. Исходные данные (количество саккад) нанесены на верхние оси; каждое среднее различие нанесено на нижние оси как распределение выборки начальной загрузки (было взято 5000 выборок начальной загрузки; доверительный интервал скорректирован на систематическую ошибку и ускорен). Средние различия обозначены точками; 95% доверительные интервалы обозначены концами вертикальных полос погрешностей.
Сосредоточившись на результатах отслеживания эллипса, с помощью апостериорного анализа мы обнаружили, что для задачи быстрого отслеживания эллипса по часовой стрелке наблюдается значительное увеличение количества саккад в группе людей с нарушением слуха по сравнению с контрольной группой ( M diff = 7,38 [95% CI 2,762, 13,736], Mann Whitney U = 32, p = 0,002, Hedge’s g = 1,02 [95% CI 222, 1,62). Точно так же мы обнаруживаем аналогичное увеличение количества саккад для группы людей с нарушением слуха при медленном движении против часовой стрелки ( M diff = 5.435 [95% ДИ 2,021, 9,6], Манна Уитни U = 34, p = 0,003, Hedges ’ г = 1,09 [95% ДИ = 0,337, 1,82]).
Обсуждение
В настоящем исследовании движения глаз тестировались в группе лиц с нарушениями слуха, глухих с раннего младенчества, с использованием задания преследования. Результаты показывают, что участники с нарушением слуха были менее точны в поддержании целевой фиксации, чем контрольная группа, в более сложных движениях в эллиптических задачах. Тем не менее, участники с нарушением слуха выполняли то же самое, что и органы управления в задачах линейного движения.Результаты также показывают, что по сравнению с контрольной группой с нормальной функцией слуха, люди с нарушением слуха демонстрируют пониженную способность отслеживать цель в эллиптических условиях и совершают больше саккад за испытание в вертикальном, против часовой стрелки и в горизонтальном направлении. в меньшей степени, эллиптические условия по часовой стрелке на обеих испытанных скоростях. Эти результаты предполагают, что глухота влияет на развитие или поддержание явного глазодвигательного поведения.
Несколько исследований показывают, что лишение сенсорной модальности может изменить развитие других модальностей (Bavelier and Neville, 2002).Церебральная пластичность после деафференцировки может привести к адаптивным или дезадаптивным изменениям поведения (Merabet and Pascual-Leone, 2010). Несколько исследований показали, что у глухих людей лучше проявляются некоторые зрительные способности, в том числе визуальное обнаружение на периферии (см. Резюме в Bavelier et al., 2006). Однако такое конкретное улучшение производительности не было обнаружено в других сенсорных системах или процессах. Что касается двигательной системы, слуховая депривация, по-видимому, приводит к неадаптивным изменениям поведения.Действительно, глухота обычно приводит к снижению общей динамической координации, равновесия, ловли мяча, времени реакции, скорости выполнения движений и моторного обучения (например, Wiegersma and Velde, 1983; Gayle and Pohlman, 1990; Siegel et al., 1991; Hartman et al., 2011). Наши результаты согласуются с общим представлением о том, что слух играет важную роль в возникновении и поддержании моторной обработки.
Поскольку движения глаз во многих отношениях проще других движений, глазодвигательная система предоставляет идеальную возможность исследовать механизмы мозга, лежащие в основе движений, управляемых визуально (Lisberger et al., 1987). Тем не менее, несмотря на относительно большое количество исследований, большинство результатов противоречивы, и неясно, как, например, слуховая депривация влияет на зрительную ориентацию (обзор см. Dye and Bavelier, 2013). Некоторые исследования показывают, что при одновременной стимуляции зрительной периферии и центрального поля зрения глухие люди могут более эффективно распределять свои ресурсы зрительного внимания в отличие от контрольных участников (Dye et al., 2009). Многие эксперименты показали, что глухие люди легче отвлекаются на ненужные отвлекающие элементы, особенно когда эти элементы появляются в поле периферического зрения (Proksch and Bavelier, 2002; Chen et al., 2006). Все эти исследования были направлены на изучение сложных процессов внимания или погони за зрительными стимулами на периферии и оценивали исключительно зрительную ориентацию внимания, не обращая внимания на то, может ли открытое глазодвигательное поведение также измениться у глухих.
Удивительно, но скрытый аспект визуального отбора зрительного глазодвигательного поведения был исследован дальше, чем открытый аспект. В исследовании Bottari et al. (2012) — единственный на сегодняшний день, кто измерил работу глаз с помощью классического открытого задания на глазодвигательное поведение — про- и антисаккад.Результаты этого исследования предполагают возможное изменение баланса между произвольной и рефлексивной ориентацией на движения глаз, более короткие латентные периоды саккад и меньшая частота ошибок были обнаружены в исследованиях просаккад, а не в исследованиях против саккад в обеих популяциях. Однако эффект был значительно сильнее у глухих, чем у слышащих участников. Наши результаты подтвердили результаты Bottari et al. (2012), предполагая, что генерация движений у глухих изменена, но мы расширили последнее, предположив, что объединенные петли визуальной и моторной обратной связи, а также контроль обратной связи, который можно оценить с помощью плавного преследования (Lizak et al., 2016), у этой популяции также нарушена. Взятые вместе, эти данные предполагают, что ранний слуховой сигнал важен для нормального развития механизмов, лежащих в основе контроля движений глаз.
При интерпретации настоящих результатов следует принимать во внимание однородность группы, представленной в этом исследовании, с точки зрения тяжести, возраста начала, прогрессирования и этиологии. У всех участников была тяжелая или глубокая потеря слуха (подробности см. В Таблице 1). У большинства участников было нарушение слуха с рождения, и только трое из них потеряли слух до начала изучения языка, а именно в возрасте от 2 до 24 месяцев.Наконец, все, кроме одного, использовали слуховые аппараты для усиления звука. Здесь у всех участников группы было одинаковое начало потери слуха, продолжительность потери слуха, использование слухового аппарата и способы общения — факторы, которые, как было выявлено, критически влияют на пластичность и поведение глухих (например, Kral and Sharma, 2012 ). Следует отметить, что общая дисперсия их результатов довольно мала и не отличается от результатов контрольной группы. Многие характеристики потери слуха следует изучить дополнительно, чтобы выявить, какие особенности вызывают больше изменений глазодвигательного поведения.В будущих исследованиях необходимо изучить влияние этих характеристик, вид поведенческих изменений, а также наличие критического периода, в течение которого слуховой сигнал необходим для развития типичного глазодвигательного поведения.
Наконец, вестибулярная функция также может быть обсуждена в связи с данными. Действительно, у значительной части людей с врожденной глухотой имеется сопутствующее вестибулярное нарушение (Buchman et al., 2004). Вестибулярные клетки участвуют в вестибулярно-окулярных рефлексах, которые позволяют нам удерживать изображения на сетчатке во время коротких движений головы (Müri and Nyffeler, 2008).В нашем исследовании у участников была зафиксирована голова, поэтому вестибулоокулярные рефлексы не генерировались во время эксперимента. Однако нейроны вестибулярного ядра также участвуют в передаче сигналов о скорости глаз при плавном преследовании (Katz, 2002; Krauzlis, 2004), поэтому они были задействованы в задаче преследования. Дальнейшие исследования потребуют определения точного воздействия слуховой депривации на глазодвигательное поведение путем контроля вестибулярных нарушений.
Заявление о доступности данныхНаборы данных, созданные для этого исследования, доступны по запросу соответствующему автору.
Заявление об этикеИсследования с участием людей были рассмотрены и одобрены всеми участниками, являвшимися добровольными добровольцами, и лечились в соответствии с Политическим заявлением Tri-Council: этическое поведение для исследований с участием людей (Совет по медицинским исследованиям Канады, MRRC, 2003). Пациенты / участники предоставили письменное информированное согласие на участие в этом исследовании.
Авторские взносы
AS, CT и DE разработали и выполнили эксперимент.Все авторы написали рукопись и обсудили результаты.
Финансирование
Это исследование финансировалось NSERC и грантом CFI для DE, грантом NSERC для AJ и европейским грантом NEST Pathfinder PERCEPT 043261 для SP.
Конфликт интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могут быть истолкованы как потенциальный конфликт интересов.
Список литературы
Баайен, Р.Х., Дэвидсон Д. Дж. И Бейтс Д. М. (2008). Моделирование смешанных эффектов с перекрещенными случайными эффектами для предметов и предметов. J. Mem. Lang. 59, 390–412. DOI: 10.1016 / j.jml.2007.12.005
CrossRef Полный текст | Google Scholar
Бухман, К. А., Джой, Дж., Ходжес, А., Телиски, Ф. Ф., и Балкани, Т. Дж. (2004). Вестибулярные эффекты кохлеарной имплантации. Ларингоскоп 114, 1–22.
Google Scholar
Чен, К., Чжан, М., и Чжоу, X.(2006). Влияние пространственного распределения внимания во время торможения возврата (IOR) на фланкерное вмешательство у слышащих и врожденно глухих людей. Brain Res. 1109, 117–127. DOI: 10.1016 / j.brainres.2006.06.043
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Дай М. В., Бавелье Д. (2013). «Визуальное внимание глухих: перспектива нейропластичности», Глухота. Справочник Springer по слуховым исследованиям , ред. А. Крал, А. Поппер и Р.Фэй (Нью-Йорк, штат Нью-Йорк: Springer), 237–263. DOI: 10.1007 / 2506_2013_9
CrossRef Полный текст | Google Scholar
Дай М. В., Хаузер П. К. и Бавелье Д. (2009). Усилено или недостаточно зрительное избирательное внимание у глухих? Корпус полезного поля зрения. PLoS One 4: e5640. DOI: 10.1371 / journal.pone.0005640
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Гейл, Г. У., и Полман, Р. Л. (1990). Сравнительное исследование динамического, статического и вращательного баланса глухих и слышащих детей. Восприятие. Mot. Навыки 70, 883–888. DOI: 10.2466 / pms.1990.70.3.883
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Hartman, E., Houwen, S., and Visscher, C. (2011). Показатели моторики и занятия спортом у глухих младших школьников. Адапт. Phys. Activ. Q. 28, 132–145. DOI: 10.1123 / apaq.28.2.132
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Heimler, B., van Zoest, W., Baruffaldi, F., Донк, М., Ринальди, П., Казелли, М.С. и др. (2015). Нахождение баланса между захватом и контролем: глазодвигательный отбор у ранних глухих взрослых. Brain Cogn. 96, 12–27. DOI: 10.1016 / j.bandc.2015.03.001
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Джаяраман С., Кляйн Р. М., Хилчи М. Д., Патил Г. С. и Мишра Р. К. (2016). Пространственные градиенты глазодвигательного торможения возврата у глухих и нормальных взрослых. Exp. Brain Res. 234, 323–330.DOI: 10.1007 / s00221-015-4439-x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Кац, Дж. (2002). Hanbook of Clinical Audiology , 5th Edn, Philadelphia: Lippincott William & Wilkins.
Google Scholar
Керзель Д., Борн С. и Соуто Д. (2010). Подавление устойчивого плавного преследования и догоняющих саккад резкими визуальными и слуховыми сигналами. J. Neurophysiol. 104, 2573–2585. DOI: 10.1152 / jn.00193.2010
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ландри, С.П., Пейдж, С., Шиллер, Д. М., Лепаж, Дж. Ф., Теорет, Х. и Шампу, Ф. (2015). Слуховые образы заставляют двигательные действия. Нейроотчет 26, 101–106. DOI: 10.1097 / WNR.0000000000000307
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ли, Р. Дж., И Зи, Д. С. (2015). Неврология движения глаз. Оксфорд: Издательство Оксфордского университета.
Google Scholar
Лисбергер, С. Г., Моррис, Э. Дж., И Тайксен, Л. (1987). Обработка визуальных движений и сенсорно-моторная интеграция для плавного отслеживания движений глаз. Annu. Rev. Neurosci. 10, 97–129. DOI: 10.1146 / annurev.ne.10.030187.000525
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Лизак Н., Клаф М., Миллист Л., Калинчик Т., Уайт О. Б. и Филдинг Дж. (2016). нарушение плавного преследования как маркер раннего рассеянного склероза. Фронт. Neurol. 7: 206. DOI: 10.3389 / fneur.2016.00206
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Мэддокс, Р. К., Посписил, Д.А., Стекер Г. К., Ли А. К. (2014). Направление взгляда усиливает различение слуховых пространственных сигналов. Curr. Биол. 24, 748–752. DOI: 10.1016 / j.cub.2014.02.021
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Мартинес-Конде, С., Макник, С. Л., Тронкосо, X. Г., и Дьяр, Т. А. (2006). Микросаккады противодействуют потускнению зрения во время фиксации. Нейрон 49, 297–305. DOI: 10.1016 / j.neuron.2005.11.033
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Муньос, Д.П. и Коу Б.С. (2011). Саккада, поиск и ориентация — нейронный контроль саккадических движений глаз. Eur. J. Neurosci. 34, 176–176. DOI: 10.1111 / j.1460-9568.2011.07810.x
CrossRef Полный текст | Google Scholar
Мюри, Р. М., и Найфелер, Т. (2008). Нейрофизиология и нейроанатомия рефлексивных и волевых саккад, выявленные исследованиями поражений у неврологических пациентов и транскраниальной магнитной стимуляцией (ТМС). Brain Cogn. 68, 284–292. DOI: 10.1016 / j.bandc.2008.08.018
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Паулсен, Дж., И Эверцен, Х. У. (1966). Аудиовизуальный рефлекс. Acta Oto Laryngol. Дополнение 224, 217–221.
Google Scholar
Прасад, С. Г., Патил, Г. С., Мишра, Р. К. (2015). Влияние экзогенных сигналов на скрытую пространственную ориентацию у глухих и нормально слышащих людей. PLoS One 10: e0141324. DOI: 10.1371 / journal.pone.0141324
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Роберт, М.П., Ингстер-Моати, И., Альбуиссон, Э., Каброл, Д., Голсе, Б., и Вайвр-Дуре, Л. (2014). Вертикальные и горизонтальные плавные следящие движения глаз у детей с нарушением координации развития. Dev. Med. Детский Neurol. 56, 595–600. DOI: 10.1111 / dmcn.12384
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Рольфс М., Энгберт Р. и Клигл Р. (2005). Кроссмодальное соединение глазодвигательного контроля и пространственного внимания в зрении и слухе. Exp.Brain Res. 166, 427–439. DOI: 10.1007 / s00221-005-2382-y
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Спаркс, Д. Л., Мэйс, Л. Е. (1990). Сигнальные преобразования, необходимые для генерации саккадических движений глаз. Ann. Rev. Neurosci. 13, 309–336. DOI: 10.1146 / annurev.ne.13.030190.001521
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Стамп, Д. М. (1993). Эвристическая фильтрация и надежные методы калибровки для систем слежения за учениками на основе видео. Behav. Res. Методы Instrum. Comput. 25, 137–142. DOI: 10.3758 / bf03204486
CrossRef Полный текст | Google Scholar
Ван Гроотель, Т. Дж., И Ван Опсталь, А. Дж. (2009). Поведение человека по локализации звука после многократных изменений положения глаз. Eur. J. Neurosci. 29, 2233–2246. DOI: 10.1111 / j.1460-9568.2009.06761.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Вигерсма, П. Х., и Велде, А. В. (1983). Двигательное развитие глухих детей. J. Child Psychol. Психиатр. 24, 103–111.
Google Scholar
Юваль-Гринберг, С., Деуэлл, Л. Ю. (2011). Записанные на кожу головы индуцированные ответы гамма-диапазона на слуховую стимуляцию и его корреляцию с активностью саккадических мышц. Brain Topogr. 24, 30–39. DOI: 10.1007 / s10548-010-0157-7
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Зан, Дж. Р., Абель, Л. А., и Делл’Оссо, Л. Ф. (1978). Аудиоокулярные характеристики отклика. Sen. Process. 2, 32–37.
PubMed Аннотация | Google Scholar
Zambarbieri, D., Schmid, R., Magenes, G., and Prablanc, C. (1982). Саккадические реакции, вызванные представлением зрительных и слуховых целей. Exp. Brain Res. 47, 417–427.
PubMed Аннотация | Google Scholar
Генная терапия восстанавливает врожденную глухоту
Источник: PAD Питтсбургская ассоциация глухих, Inc.Международная исследовательская группа, возглавляемая учеными из Франции, использовала генную терапию для восстановления слуха у взрослых мышей с глухотой по DFNB9, одной из наиболее распространенных форм наследственной или врожденной глухоты.Помимо предотвращения потери слуха при введении в развивающуюся улитку молодых мышей, подход генной терапии — при котором отдельные вирусные векторы используются для доставки белка отоферлина в виде двух половинок — также обращает вспять глухоту у взрослых мышей DFNB9.
Ученые говорят, что их достижение может проложить путь к испытаниям генной терапии на людях у пациентов с глухотой по DFNB9. «Наши результаты подтверждают как профилактическую, так и лечебную эффективность местной генной терапии на мышиной модели DFNB9, одновременно расширяя сферу потенциальных применений генной терапии AAV для форм наследственной глухоты человека», — написали они в своей опубликованной статье в PNAS , которая озаглавлен: «Генная терапия, опосредованная двойным AAV, восстанавливает слух у мышей DFNB9.В состав группы вошли исследователи из Института Пастера, Инсерма, CNRS, Коллеж де Франс, Университета Сорбонны, Университета Клермон Овернь во Франции, а также коллег из Университета Флориды, Гейнсвилля, Медицинского центра Колумбийского университета и пресвитерианской церкви Нью-Йорка. Больница в США
Аутосомно-рецессивные генетические формы глухоты являются причиной большинства случаев глубокой потери слуха, пишут исследователи. «Более половины случаев несиндромальной глубокой врожденной глухоты имеют генетическую причину, и большинство (~ 80%) являются аутосомно-рецессивными (DFNB).«Кохлеарные имплантаты в настоящее время являются единственным вариантом лечения, и, хотя они могут помочь улучшить слух, они не идеальны. У пациентов по-прежнему часто возникают проблемы с распознаванием речи в шумной обстановке или, например, при прослушивании музыки.
Аденоассоциированные вирусы (AAV) являются многообещающими векторами для доставки генов в генной терапии и могут быть использованы для доставки генов, необходимых для восстановления слуха при врожденных формах глухоты. Устойчивая экспрессия трансгена с AAV была достигнута на различных моделях животных, а превосходный профиль безопасности этих векторов был продемонстрирован во многих продолжающихся клинических испытаниях », — отметили авторы.«В результате AAV теперь являются основной системой доставки генов, используемой в генной терапии генетических заболеваний».
Однако в большинстве тестов на моделях глухоты на мышах требуемый ген вводили в развивающуюся улитку новорожденных мышей (обычно примерно на 1 или 2 день после рождения — P1 или P2), то есть до того, как животные начинают слышать. , около P12. Напротив, у человека развитие улитки завершается у плода во время беременности, и слух становится возможным примерно к 20 неделе беременности, задолго до рождения.Это приводит к очень маленькому терапевтическому окну, если генная терапия должна проводиться в развивающейся улитке. В действительности генную терапию необходимо проводить после рождения, после того, как потеря слуха и ее причина будут диагностированы у младенца, и после того, как улитка полностью разовьется. Это означает, что лечение должно иметь возможность обратить вспять ранее существовавшую глухоту, и это следует учитывать при разработке доклинических тестов.
Левая панель представляет собой схематическое изображение человеческого уха.Звуковые волны собираются наружным ухом, состоящим из ушной раковины и слухового прохода. Среднее ухо, состоящее из барабанной перепонки и косточек, передает звуковые волны во внутреннее ухо, которое представляет собой улитку — орган слуха, ответственный за передачу слуховых сообщений в центральную нервную систему. На правой панели показано иммунофлуоресцентное изображение слухового сенсорного эпителия внутри улитки после инъекции. Внутренние волосковые клетки окрашены на отоферлин в зеленый цвет. Отоферлин обнаруживается почти во всех этих клетках.Вставка представляет собой область с большим увеличением, показывающую внутреннюю волосковую клетку, которая не была преобразована. [© Institut Pasteur] Чтобы протестировать свой подход к генной терапии против уже существовавшей глухоты, исследователи во главе с Саидом Сафиддином, доктором философии, исследователем CNRS в области генетики и физиологии слухового аппарата (Institut Pasteur / Inserm), обратились к мышиной модели. DFNB9, который является причиной 2-8% врожденной глухоты у людей. Заболевание вызвано мутациями в гене, кодирующем отоферлин, белок, который играет роль в передаче звуковой информации в синапсах внутренних волосковых клеток (IHC) в улитке.У этих мышей отсутствует ген отоферлина (Otof — / — животных).
Потенциальное использование технологии AAV для доставки гена отоферлина должно быть затруднено из-за ограниченной способности ДНК-упаковки векторов AAV — около 4,7 т.п.н. ДНК — чего было бы недостаточно для переноса полного рекомбинантного гена отоферлина, который составляет около 6 т.п.н. Команда Саффидина разработала способ обойти это, используя подход с двойным AAV-вектором, в котором ген отоферлина эффективно разделяется между двумя разными векторами AAV, каждый из которых кодирует один из двух противоположных концов гена отоферлина.
Команда сначала одновременно вводила два вектора непосредственно в улитку мышей DFNB9, у которых отсутствует ген отоферлина, на 10-й день постнатального развития (P10). Это происходит до завершения развития улитки. Анализ ткани улитки через восемь недель после однократной инъекции обоих векторов подтвердил, что обработка приводила к продукции полного белка отоферлина более чем в 60% ИГК. Тесты слуховой реакции ствола мозга через 4 недели после доставки гена подтвердили, что слух у животных был восстановлен, и это восстановление слуха все еще было очевидным у большинства мышей более чем через 6 месяцев.«Через тридцать недель после инъекции у шести из восьми мышей, получавших инъекции P10, пороги слышимости все еще были в пределах 10 дБ от таковых у мышей дикого типа», — пишут авторы. «Таким образом, генная терапия до появления слуха предотвращает глухоту у мышей Otof — / — .
Обнадеживает то, что тесты на мышах, получавших однократную интракухлеарную инъекцию векторной пары на P17 или P30, показали, что лечение приводило к продукции отоферлина в IHC во всей обработанной улитке и приводило к восстановлению слуха уже через 3 недели после инъекции. до 20 недель, когда животных снова тестировали.
«Мы сообщаем здесь, в модели мышей DFNB9, наше доказательство принципа того, что кохлеарная доставка фрагментированной кДНК с помощью подхода с двумя векторами AAV может эффективно восстанавливать производство полноразмерного белка, что приводит к долговременной коррекции фенотип глубокой глухоты у этих мышей », — заключили авторы. «Мы показываем, что местная генная терапия у мутантных мышей не только предотвращает глухоту при введении в незрелые органы слуха, но также надолго восстанавливает слух при введении на зрелой стадии, что вселяет надежды на будущие испытания генной терапии у пациентов с DFNB9.Результаты также показывают, что терапевтическое окно для генной терапии против глухоты DFNB9 может быть дольше, чем считалось ранее, «вселяя надежду на будущие испытания генной терапии у пациентов с DFNB9».
.