Сравнение препаратов для повышения потенции
Большинство мужчин с возрастом (а некоторые и раньше) начинают испытывать трудности с потенцией и приходят к самому очевидному и эффективному решению – купить таблетки. Перед тем, как выбрать препарат для потенции, стоит изучить предлагаемые фармацевтиками средства. Ассортимент их очень широк, а условия применения различаются практически по всем параметрам.
Типы препаратов для повышения потенции
Для лечения эректильной дисфункции (ЭД) в традиционной медицине используется несколько групп препаратов. Выбор зависит от диагностированной причины появившихся проблем и механизма действия:
- Средства, повышающие уровень оксида азота: ингибиторы ФДЭ-5 (Виагра, Сиалис, Левитра), активаторы NO-синтазы (Импаза).
- Альфа-адреноблокаторы: Йохимбин, Фентоламин.
- Аналоги простагландина Е: Алпростадил.
- Андрогены: препараты тестостерона, Метилтестостерон, Андриол, Местеролон.

Не зная, какой препарат для потенции выбрать, некоторые мужчины начинают с применения народных средств, БАД-ов, специальных упражнений и дыхательных техник. Эффективность таких методов сомнительна, а в некоторых случаях полностью отсутствует. Между тем проблема может требовать быстрого решения – в случае патологической причины ЭД.
Ингибиторы фосфодиэстеразы 5 типа, особенности
Препараты этой группы считаются наиболее безопасными и эффективными – это доказано использованием их во всем мире уже более 20 лет. Миллионы мужчин получили возможность быть уверенными в себе и иметь полноценный секс, просто приняв таблетку.
Действие ингибиторов ФДЭ-5 заключается в приливе крови к половым органам и замедлении венозного оттока. Происходит это после нескольких биохимических реакций и расслабления гладкой мускулатуры кавернозных тел пениса. Еще одна особенность – такой эффект наблюдается только при выраженной сексуальной стимуляции.
Помимо этого, есть и другие положительные моменты:
- Препараты благотворно влияют на работу сердечно-сосудистой системы.
- Ингибиторы ФДЭ-5 можно использовать как для единичного секса, так и при регулярной половой жизни.
- Средства этой группы сохраняют эффект от 4-5 до 36 ч. – их применяют не только как разовый способ повышения потенции, но и при постоянной терапии.
При сравнении препаратов для повышения потенции из разных групп видно, что ингибиторы ФДЭ-5 – наиболее популярные у мужской половины человечества средства. За изобретение Виагры ученым-медикам даже была присуждена Нобелевская премия.
Сравнение Виагры, Сиалиса и Левитры
У всех трех препаратов активное вещество – из группы ингибиторов ФДЭ-5. Однако свойства уже готовых таблеток различны:
- Виагра (силденафил). Самое мощное средство с доступной ценой и широким ассортиментом дженериков. Кроме того, выпускается в форме геля, капсул, софт и даже шипучки. Эффект наступает примерно через 1 ч. и длится до 4-6 ч. – в зависимости от особенностей здоровья, массы тела, употребления жирной пищи.
- Сиалис (тадалафил). Благодаря слегка отличному химическому составу действует на протяжении 36 ч. Можно принять таблетку с утра и быть готовым к сексу в любое время. Как и Виагру, его принимают 1 р. в сут., однако рекомендованная доза в 2,5 р. меньше – всего 20 мг. Из всех дженериков имеет самую низкую цену и применяется даже в терапии эректильной дисфункции.
- Левитра (варденафил). Фармакологическое действие примерно такое же, как у Виагры: эффект наступает через 40-60 мин. и длится до 6 ч. Однако именно Левитру предпочитают многие пожилые пациенты: немного уступая Виагре в мощности, она почти не имеет побочных эффектов, в то время как рекомендованная дозировка та же, что и у Сиалиса – всего 20 мг.
 Благодаря слегка отличному химическому составу действует на протяжении 36 ч. Можно принять таблетку с утра и быть готовым к сексу в любое время. Как и Виагру, его принимают 1 р. в сут., однако рекомендованная доза в 2,5 р. меньше – всего 20 мг. Из всех дженериков имеет самую низкую цену и применяется даже в терапии эректильной дисфункции.
Благодаря слегка отличному химическому составу действует на протяжении 36 ч. Можно принять таблетку с утра и быть готовым к сексу в любое время. Как и Виагру, его принимают 1 р. в сут., однако рекомендованная доза в 2,5 р. меньше – всего 20 мг. Из всех дженериков имеет самую низкую цену и применяется даже в терапии эректильной дисфункции.Сравнительная характеристика препаратов для потенции позволяет выбрать самое эффективное средство в каждом конкретном случае – с учетом индивидуальных запросов и возможностей здоровья.
Особенности медикаментозного лечения импотенции
Зачастую в развитии импотенции виновата не одна причина, а целый комплекс: основная определяющая с букетом провоцирующих факторов. Это могут быть нарушения неврологического и психологического характера, отягощенные гормональными, анатомическими, сосудистыми патологиями. Поставить правильный диагноз и грамотно составить схему лечения в большинстве случаев доступно только специалисту. Он сделает обзор препаратов для повышения потенции и порекомендует самое эффективное средство для вашего конкретного случая.
Это могут быть нарушения неврологического и психологического характера, отягощенные гормональными, анатомическими, сосудистыми патологиями. Поставить правильный диагноз и грамотно составить схему лечения в большинстве случаев доступно только специалисту. Он сделает обзор препаратов для повышения потенции и порекомендует самое эффективное средство для вашего конкретного случая.
Если проблема находится на начальной стадии, можно попробовать дженерики препаратов из группы ингибиторов ФДЭ-5 – они достаточно безопасны и дают гарантированный результат. Как и любые медикаменты, у них есть список особых условий – перед применением обязательно изучите инструкцию.
АКТИВАТОРЫ ПРЕКОНДИЦИОНИРОВАНИЯ МИОКАРДА – НОВЫЕ ВОЗМОЖНОСТИ ЛЕЧЕНИЯ ИБС?
Farkos.UA » Публікації » АДВОКАРД » АКТИВАТОРЫ ПРЕКОНДИЦИОНИРОВАНИЯ МИОКАРДА – НОВЫЕ ВОЗМОЖНОСТИ ЛЕЧЕНИЯ ИБС?
В последние годы в рекомендации Рабочей группы Европейского общества кардиологов по ведению больных стабильной стенокардией (2006) в качестве базисной терапии введен класс активаторов калиевых каналов. Именно группа активаторов калиевых каналов стала объектом всестороннего изучения, поскольку представляет интерес в связи с возможностью терапевтического влияния на процессы, направленные на формирование особого состояния миокарда — прекондиционирования.
Именно группа активаторов калиевых каналов стала объектом всестороннего изучения, поскольку представляет интерес в связи с возможностью терапевтического влияния на процессы, направленные на формирование особого состояния миокарда — прекондиционирования.
Развитие прекондиционирования миокарда приводит к значительному ограничению зоны инфаркта, уменьшению ультраструктурных повреждений, снижению частоты развития аритмий. Процесс прекондиционирования протекает в 2 стадии: ранняя (во время которой запускается каскадный медиаторный механизм, приводя к развитию ишемического прекондиционирования) и поздняя — «второе окно защиты» — повышает толерантность в отношении летальной ишемии миокарда через 24 ч. длительностью до 72 ч. В механизмах отдаленного прекондиционирования первостепенное значение имеет активация А1-рецептора аденозина, модулирующее влияние системы, генерирующей NO, и, связанной с этим, активации синтезов белков теплового шока (HSP — heat shock protein).
Оксид азота (NO) в развитии отдаленного (позднего) ишемического прекондиционирования выступает модулятором механизмов его развития. При этом различные изоформы NO-синтазы (NOS) имеют различное назначение: зависимая от кальция эндотелиальная (eNOS), инициирует ишемическое прекондиционирование в ранней фазе, и независимая от кальция индуцибельная (iNOS), генерирующая NO определяет развитие защиты миокарда против ишемии во второй день.
При этом различные изоформы NO-синтазы (NOS) имеют различное назначение: зависимая от кальция эндотелиальная (eNOS), инициирует ишемическое прекондиционирование в ранней фазе, и независимая от кальция индуцибельная (iNOS), генерирующая NO определяет развитие защиты миокарда против ишемии во второй день.
В исследовании показано, что дробное повторное внутривенное введение аденозина «вводит» миокард в состояние резистентности к ишемии, сравнимое по эффективности с действием второго защитного окна и длящееся несколько недель.
Эти данные подтверждаются с позиций «доказательной медицины». В клинических трайлах AMISTAD I (1999) и AMISTAD II (2005) аденозин в достаточной дозе на 57% уменьшал размер инфаркта у больных с передним инфарктом миокарда. Кроме того, аденозин показал себя перспективным как дополнение к кардиоплегии.
Недостатком естественного активатора А1-рецепторов аденозина, следует считать неудобство применения, связанное с необходимостью внутривенного введения через инфузомат под тщательным контролем показателей гемодинамики, а также быстрой деградацией препарата в кровеносном русле. Поэтому, перспективным направлением является создание веществ, подобных аденозину, но имеющих благоприятный фармакокинетический профиль. Именно такой является субстанция магладена — аденозин-трифосфато-Мд(11)-глюконат, входящая в состав комбинированного препарата Адвокард® производства фармацевтической фирмы «ФарКоС» (Украина).
Терапевтическая активность Адвокарда® обусловлена активацией пуриновых (аденозиновых) рецепторов, стимуляцией АТФ-чувствительных калиевых каналов под воздействием магладена-аденозин-5′-трифосфатоглюконато-магния (II) тринатриевой соли. Это угнетает вхождение ионов кальция в клетки, что проявляется антиишемическим, мембраностабилизирующим действием и антиаритмическим эффектом.
В состав Адвокарда® входит мол-сидомин, который выступает в роли модулятора (а не донатора) эффектов NО, связанных со стимуляцией процессов прекондиционирования активацией аденозиновых рецепторов магладеном. Таким образом, Адвокард® проявляет, в первую очередь, свойство цитопротектора, а уже, во-вторых, оказывает положительное влияние на показатели системной гемодинамики: стимуляция пуриновых рецепторов сопровождается дилатацией мелких артериол, а мол-сидомин расширяет вены среднего диаметра, поэтому такое потенциирование является гемодинамически выгодным для организма.
Повний текст статті ви можете скачати у форматі PDF
Аналитика — научно-технический журнал — Аналитика
Получен комплексный нуклеотидный препарат из дрожжей Saccharomyces cerevisiae раса 14. Определены оптимальные условия для выделения продуктов аутоферментативного гидролиза нуклеиновых кислот. Проанализирован состав биологически активных веществ препарата и выявлено содержание адениновых нуклеотидов, а также других биологически активных веществ: аминокислот, витаминов, микро- и макроэлементов, жирных кислот, в том числе полиненасыщенных. Проведена оценка активирующего действия нуклеотидного препарата на Са2+-зависимую NO-синтазу по сравнению с Т-активином. Приведена схема автоматизированной установки для получения субстанции.
Проведена оценка активирующего действия нуклеотидного препарата на Са2+-зависимую NO-синтазу по сравнению с Т-активином. Приведена схема автоматизированной установки для получения субстанции.Известно, что Ca2+-зависимая NO-синтаза является одним из ключевых ферментов антиоксидантной защиты организма [1]. Снижение ее активности сопровождает возникновение и развитие таких заболеваний, как диабет, атеросклероз, инфаркт миокарда, сердечная недостаточность [2, 3]. В этой связи особую актуальность приобретает поиск активаторов Ca2+-зависимой NO-синтазы (NOS). Среди них наиболее перспективны для использования в качестве лекарственных средств производные адениннуклеотидов, полученные из различных биологических объектов, в частности из дрожжей Saccharomyces cerevisiae [4].
ОБЪЕКТЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
Объектом исследования служили штаммы дрожжей Saccharomyces cerevisiae. Для получения нуклеотидного препарата использованы пять штаммов дрожжей Saccharomyces cerevisiae раса 14, которые выращивали на питательной среде следующего состава:
• макроэлементы, г/л:
Kh3PO4 – 1,0;
K2HPO4 – 0,13;
MgSO4·7h3O – 0,7;
NaCl – 0,1;
(Nh5)2SO4 – 3,5;
• микроэлементы, мг/л:
ZnSO4 ·6h3O – 0,18;
CuSO4·5h3O – 0,15;
MnSO4·5h3O – 0,15;
CoCl2 – 0,18;
• витамины, мг/л: тиамин – 0,4; биотин – 0,1;
• источник углеродного питания глюкоза – 10,0 г/л.
Активность Са2+-зависимой NOS в тимоцитах крыс Wistar определяли по методу [5] с некоторыми модификациями. В основу положено измерение активности NOS по конечному продукту NO, который после перевода в NO2– количественно определяется с помощью реактива Грисса. Реактив Грисса готовили из сульфаниловой кислоты и α-нафтилэтиламина, взятых в соотношении 1 : 1 (по массе) с последующим растворением 10 г смеси в 90 мл 12%-ной уксусной кислоты.
К 800 мкл полученного супернатанта (тимоцитов) добавляли в строго определенном порядке: сначала 100 мкл 20 мМ K2-фосфатного буфера рН = 7,4; затем – 50 мкл L-аргинина и в конце – 50 мкл 5,4 · 10–4 М NADPH (Sigma, США). NADPH “запускает” ферментную реакцию, поэтому нарушение последовательности внесения в смесь реактивов может приводить к заниженным результатам [5]. Кинетические параметры ферментативных реакций рассчитывали по уравнению Михаэлиса – Ментен с использованием пакета программ SPLab4. Определяли значение кажущейся константы Михаэлиса (Kmapp) и удельную скорость (vуд) NOS-катализируемой реакции [6].
Подготовленную реакционную смесь инкубировали на водяной бане с вибрацией при температуре 37 °C в течение 10 мин, после чего реакцию останавливали добавлением 300 мкл реактива Грисса (Sigma). Пробы извлекали из водяной бани и оставляли при комнатной температуре (20 °C) на 30 мин для максимального развития окраски [6].
Концентрацию NO2– определяли на спектрофотометре Beckman DU-65 (США) при длине волны 548 нм по интенсивности окраски фиолетово-красного азокомплекса, образовавшегося в результате реакции между сульфаниловой кислотой, NO2– и α-нафтилэтиламином. Чувствительность метода – 10–7 М NO2–.
Состав и содержание аминокислот в препарате измеряли на аминокислотном анализаторе Varian (США).
Микро- и макроэлементы определяли методом атомно-адсорбционной спектроскопии, используя спектрометр фирмы Varian (США).
Витамины, нуклеиновые кислоты и углеводы определяли с помощью жидкостного хроматографа компании Waters (США). Колонки Symmetry C18 (Waters) использовали для анализа водорастворимых витаминов (В1 – тиамин, В2 – рибофлавин, В3 – ниацин, витамин PP, В4 – холин, В5 – пантотеновая кислота, В6 – пиридоксин, В9 – фолиевая кислота, Н – биотин) и углеводов. Колонки Nucleosil C18 производства Macherey-Nagel (Германия) применяли для определения содержания жирорастворимых витаминов E (токоферол) и K (филлохинон, пренилменахинон). Жирные кислоты измеряли на обращенно-фазовой колонке C-8 (Shimadzu, Япония). Для анализа нуклеиновых кислот применяли DEAE–анионообменную колонку Nucleogen 4000-7 (Macherey-Nagel) [8].
Колонки Nucleosil C18 производства Macherey-Nagel (Германия) применяли для определения содержания жирорастворимых витаминов E (токоферол) и K (филлохинон, пренилменахинон). Жирные кислоты измеряли на обращенно-фазовой колонке C-8 (Shimadzu, Япония). Для анализа нуклеиновых кислот применяли DEAE–анионообменную колонку Nucleogen 4000-7 (Macherey-Nagel) [8].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Высокая устойчивость рибонуклеаз к воздействию денатурирующих факторов, в том числе и сильно кислой среды [8], была положена в основу разработанного нами метода денуклеинизации биомассы дрожжей [4]. Биомассу дрожжей использовали в стационарной фазе роста, когда происходило максимальное образование нуклеиновых кислот (НК) [7, 10]. Условия выбраны экспериментальным путем, контроль роста биомассы проводили микроскопически при помощи инвертированного микроскопа с камерой Nicon. На рис.1а приведена микрофотография дрожжевых колоний в конце первых суток культивирования (фото в отраженном свете). На третий день после начала выращивания прирост дрожжевых клеток достигает оптимального значения при большом числе делящихся колоний (рис. 1б – фото в поляризованном свете).
1б – фото в поляризованном свете).
Дрожжевую биомассу с концентрацией 40 г/л (по влажному весу) и рН = 6,8–7,0 при постоянном перемешивании нагревали при 90 °С в течение 20 мин. На этой стадии происходила денатурация протеолитических ферментов и рибосом, в то время как эндогенные нуклеазы сохраняли свою активность и в дальнейшем участвовали в процессе расщепления НК. Это подтверждается материалами опубликованных работ [7, 9, 10]. Затем нагревание прекращали и суспензию дрожжевой биомассы выдерживали 3 ч при 34 °С, постоянно перемешивая. На этой стадии эндогенные нуклеазы дрожжевых клеток деполимеризировали большую часть НК до олигонуклеотидов. По окончании этого времени рН среды доводили до 1,0 с помощью 1Н HCl и одновременно прогревали суспензию до 80–85 °С в течение часа. Это позволяло сделать оболочки дрожжевых клеток проницаемыми для деполимеризованных НК и других веществ, входящих в клеточный пул: витаминов, свободных внутриклеточных аминокислот, микро- и макроэлементов, углеводов и жирных кислот, кроме белков. Стадия выделения продуктов аутоферментативного гидролиза НК должна происходить при соблюдении рН 1,0–1,5 и температурного режима 80–85 °С. Об этом свидетельствуют экспериментальные данные, представленные на рис.2. Наименьшее количество нуклеиновых кислот в биомассе зафиксировано при температуре 80–90 °С (рис.2а). При рН = 1,0 через час выделяется наибольшее количество нуклеиновых кислот, а в биомассе их остаток минимален (рис.2б).
Стадия выделения продуктов аутоферментативного гидролиза НК должна происходить при соблюдении рН 1,0–1,5 и температурного режима 80–85 °С. Об этом свидетельствуют экспериментальные данные, представленные на рис.2. Наименьшее количество нуклеиновых кислот в биомассе зафиксировано при температуре 80–90 °С (рис.2а). При рН = 1,0 через час выделяется наибольшее количество нуклеиновых кислот, а в биомассе их остаток минимален (рис.2б).
После завершения процесса денуклеинизации добавляли 20–40% NaOH для доведения рН среды до 6,0–7,0; центрифугировали при 3 000 об/мин 10 мин и получали целевой продукт – комплексный нуклеотидный препарат. О полноте денуклеинизации судили по снижению НК в биомассе и наличии аденинового пула в центрифугате. Это подтверждали аналитически по спектрам поглощения образцов центрифугата, в качестве стандарта использовали аденин (Sigma). На рис.3 представлен типичный спектр поглощения адениновой компоненты в образце препарата. Абсорбционный спектр характеризуется максимумом поглощения при длине волны 257 нм и минимумом при 230 нм. Это позволяет принять 257 нм за аналитическую длину волны.
Это позволяет принять 257 нм за аналитическую длину волны.
Оценку влияния полученного образца нуклеотидного препарата на способность активировать Са2+-зависимую NOS в сравнении с иммуностимулятором Т-активином проводили на тимоцитах крыс Wistar. При этом Т-активин применяли в эффективных терапевтических дозах, а тестируемые образцы нуклеотидного препарата – в сравнимых с рекомендуемыми для нуклеината натрия количествах [10]. Эксперименты показали, что нуклеотидный препарат при трехкратном внутрибрюшинном введении дозы 10 мг/кг оказывает выраженное активирующее действие на Са2+-зависимую NOS из тимоцитов крыс (табл.1).
По степени активирующего действия нуклеотидный препарат превосходит Т-активин, о чем свидетельствует уменьшение Kmapp для препарата в 2,7 раза по сравнению с Kmapp реакции Т-активина с Са2+-зависимой NOS. Скорость NOS-катализируемой реакции выше у нуклеотидного препарата по сравнению с Т-активином (см. табл.1), то есть в рекомендованной дозе 10 мг/кг он является более сильным активатором Са2+-зависимой NOS.
В полученном нуклеотидном препарате определены некоторые биологически активные вещества: адениновые нуклеотиды, аминокислоты, витамины, микро- и макроэлементы (табл.2).
Успешные исследования по выделению нуклеотидного препарата, содержащего комплекс биологически активных веществ, а также доказательства его положительного влияния на иммунную систему, обусловили актуальность создания автоматизированного промышленного комплекса для выделения субстанции. Конструкция установки (рис.4) предполагает наличие ферментера (1) для выращивания культуры клеток, снабженного насосом (2), который соединен с центрифужным сепаратором (3) для отделения дрожжевых клеток. После сепарации дрожжевые клетки с помощью клапана (4) попадают в резервуар, который имеет открывающееся дно (7) и предфильтр (10). Резервуар снабжен паровой рубашкой (9) для нагрева до нужной температуры, а также мешалкой (8). С помощью насоса через входной патрубок в резервуар подается раствор НCl (6). После проведения описанных выше процедур полупродукт сливается через выходной патрубок (11), после чего центрифугируется.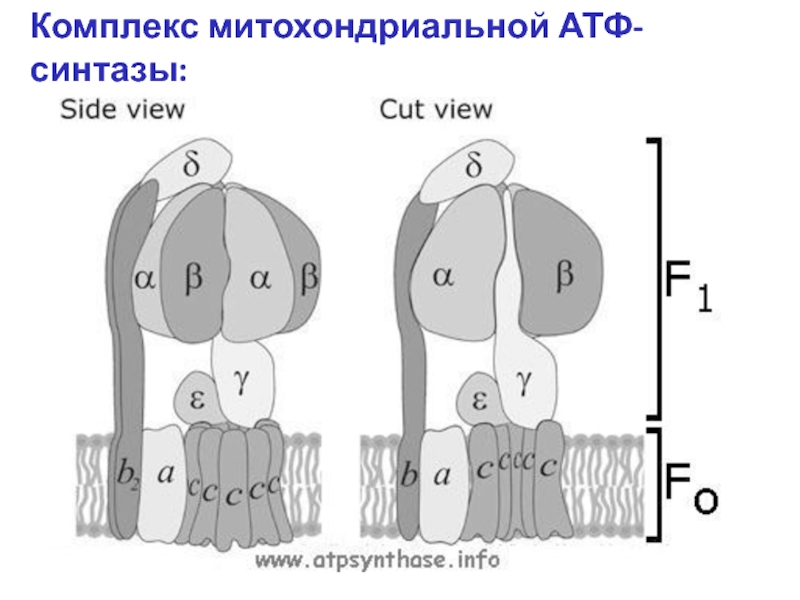 Далее центрифугат подвергают лиофильной сушке. В настоящий момент проходит этап конструирования промышленного образца установки.
Далее центрифугат подвергают лиофильной сушке. В настоящий момент проходит этап конструирования промышленного образца установки.
ЗАКЛЮЧЕНИЕ
Получен комплексный нуклеотидный препарат из дрожжей Saccharomyces cerevisiae раса 14, определено содержание адениновых нуклеотидов, а также других биологически активных веществ – аминокислот, витаминов, микро- и макроэлементов. Выявлено, что оптимальными условиями для выделения продуктов аутоферментативного гидролиза нуклеиновых кислот является рН 1,0–1,5 и температурный режим 80–85 °С.
Нуклеотидный препарат не содержит в своем составе компонентов, не соответствующих обмену веществ человека и животных. Полученная субстанция оказывает активирующее действие на Са2+-зависимую NO-синтазу, играющую важную роль в функционировании системы иммунитета и в процессах оксидативного стресса.
***
Работа выполнена при финансовой поддержке Министерства образования и науки РФ по Программе реализации комплексных проектов по созданию высокотехнологичного производства (Постановление Правительства РФ № 218 oт 09. 04.2010 г., шифр конкурса 2016-218-09). Договор
04.2010 г., шифр конкурса 2016-218-09). Договор
№ 03.G25.310258.
ЛИТЕРАТУРА:
1. Vallance P., Leone A., Calver A., Collier J., Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure . – Lancet., 1992. P. 235–240.
2. Catravas John D., Callow Allan D., Ryan Una S. Vascular Endothelium: Physiological Basis of Clinical Problems II. (NATO ASI Series A: Life Sciences Vol. 257). 1994. 230 p.
3. Chade A.R., Rodriguez-Porcel M., Herrmann J., Zhu X.,
Grande J.P., Napoli C., Lerman A., Lerman L.O. Antioxidant intervention blunts renal injury in experimental renovascular disease // J. Am. Soc. Nephrol. 2004. 15(4). P. 958–966.
4. Способ получения биомассы микроорганизмов с низким содержанием нуклеиновых кислот: пат. 2128218 РФ: МПК: C12N / Орлова В.С. [и др.]; заявители и патентообладатели: Орлова В.С., Орлова Е.В., Уваров Л. А., Евстигнеев А.А.; заявл. 31.08.1994; опубл. 27.03.1999. 5 с.
5. Hwang S. , Lopez C.A., Heck D.E., Gardner C.R., Laskin J.D. Osteopontin inhibits induction of nitric oxide synthase gene expression by Inflammatory mediators in mouse kidney epithelial cells // J. of Biol. Chem. 1994. V. 269. P. 711–715.
, Lopez C.A., Heck D.E., Gardner C.R., Laskin J.D. Osteopontin inhibits induction of nitric oxide synthase gene expression by Inflammatory mediators in mouse kidney epithelial cells // J. of Biol. Chem. 1994. V. 269. P. 711–715.
6. Bobadoe M.F., Bamisi O.O., Enujiugha V.N. Hypolipidemic and antioxidative effects of african star apple juice (chrysophylum albidum) on rats fed on diets high in cholesterol and oil // Food and Nutrition Sciences. 2016. V. 7. P. 825–843.
7. Орлова Е.В. Разработка и экспериментальное обоснование клинического применения комплексного нуклеотидного препарата из дрожжей Saccharomyces cerevisiae //
дисс… докт. биол. наук. – М., 2007. 233 с.
8. Nishiyama-Naruke A., Souza J.A., Carnelуs M., Curi R., A. HPLC determination of underivatized fatty acids saponified at 37 °C analysis of fatty acids in oils and tissues // Analytical letters. 1998. V. 31. № 14. P. 2565–2576.
9. Birkenmeier G.F., Ryan C.A. Wound signaling in tomato plants. Evidence that ABA Is not a primary signal for defense gene activation // Plant Physiol. 1998. 117(2). P. 687–693.
Evidence that ABA Is not a primary signal for defense gene activation // Plant Physiol. 1998. 117(2). P. 687–693.
10. Farron-Furstenthal F. An inhibitor protein of nuclear protein kinases // Nature (Lond.). 1979. 280. P. 415–417.
11. Машковский М.Д. Лекарственные средства : в 2-х т.: Пособие для врачей / 13-е изд. – Харьков: Торсинг, 1997. 560 с.
Способ получения селективного активатора ca2+-зависимой no-синтазы
Способ получения селективного активатора Са2+-зависимой NO-синтазы, характеризующийся тем, что дрожжи Saccharomyces cerevisiae, выращенные на синтетической среде с глюкозой, суспендируют в водопроводной воде, нагретой до 60-75°С, до концентрации 50 г сухой биомассы на 1 л, нагревают до 75-90°С и выдерживают в течение 15-25 мин при этой температуре, затем охлаждают до 35-60°С, инкубируют при этой температуре в течение 1,5-2,5 ч, после чего рН суспензии доводят до 1,0-1,5, повторно нагревают до 75-85°С и выдерживают в течение 30-60 мин, затем суспензию разводят водой в соотношении от 1:1 до 1:10 и доводят рН суспензии до 5. 0-6.0, выдерживают при помешивании 30-60 мин, после чего надосадочную жидкость центрифугированием отделяют от клеточной биомассы дрожжей и затем высушивают.
0-6.0, выдерживают при помешивании 30-60 мин, после чего надосадочную жидкость центрифугированием отделяют от клеточной биомассы дрожжей и затем высушивают.
СПОСОБ ПОЛУЧЕНИЯ СЕЛЕКТИВНОГО АКТИВАТОРА CA2+-ЗАВИСИМОЙ NOСИНТАЗЫ Изобретение относится к области биотехнологии, медицины, фармакологии, в частности к способу получения селективного активатора Са 2+-зависимой NO-синтазы (далее NOS), которая является одним из ключевых ферментов антиоксидантной защиты организма, а также эффективным регулятором Са-зависимых процессов в стволовых клетках, что особенно важно для управления функциональными механизмами стволовой клетки in vivo. Техническим результатом, на получение которого направлено изобретение, является разработка селективного активатора Са 2+-зависимой NOS, характеризующегося высокой степенью активации, а также простого способа его изготовления, отличающегося простотой изготовления и доступностью необходимого для этого сырья. Технический результат достигается в селективном активаторе Са 2+-зависимой NOS, полученном следующим образом: дрожжи Saccharomyces cerevisiae(пекарские дрожжи), выращенные на синтетической среде с глюкозой, суспендируют в водопроводной воде, нагретой до 60-75 С, до концентрации 50 г сухой биомассы на 1 л,нагревают до 75-90 С и выдерживают в течение 15-25 мин при этой температуре, затем охлаждают до 35-60 С и инкубируют при этой температуре в течение 1,5-2,5 ч. Затем рН суспензии доводят до 1,0-1,5, повторно нагревают до 75-80 С и выдерживают в течение 30-60 мин. Затем суспензию разводят водой в соотношении от 1:1 до 1:10, доводят рН до 5,0-6,0 и выдерживают при помешивании 30-60 мин. Далее надосадочную жидкость центрифугированием отделяют от клеточной биомассы дрожжей и затем высушивают.(71)(72)(73) Заявитель, изобретатель и патентовладелец: применения комплексного нуклеотидного ОРЛОВА ЕЛЕНА ВЛАДИМИРОВНА; препарата из дрожжей Saccharomyces ОРЛОВА ВАЛЕНТИНА СЕРГЕЕВНА;cerevisiae. Диссертация на соискание КЛУБКОВ ВЛАДИМИР ученой степени доктора биологических КОНСТАНТИНОВИЧ (RU) наук. — М., 2007, с. 45-47, 77-84, 94-95 Изобретение относится к области биотехнологии, медицины, фармакологии, в частности к производству селективного активатора Са 2+-зависимой NO-синтазы (далее NOS), которая является одним из ключевых ферментов антиоксидантной защиты организма, а также эффективным регулятором Сазависимых процессов в стволовых клетках, что особенно важно для управления функциональными механизмами стволовой клетки in vivo.
Затем рН суспензии доводят до 1,0-1,5, повторно нагревают до 75-80 С и выдерживают в течение 30-60 мин. Затем суспензию разводят водой в соотношении от 1:1 до 1:10, доводят рН до 5,0-6,0 и выдерживают при помешивании 30-60 мин. Далее надосадочную жидкость центрифугированием отделяют от клеточной биомассы дрожжей и затем высушивают.(71)(72)(73) Заявитель, изобретатель и патентовладелец: применения комплексного нуклеотидного ОРЛОВА ЕЛЕНА ВЛАДИМИРОВНА; препарата из дрожжей Saccharomyces ОРЛОВА ВАЛЕНТИНА СЕРГЕЕВНА;cerevisiae. Диссертация на соискание КЛУБКОВ ВЛАДИМИР ученой степени доктора биологических КОНСТАНТИНОВИЧ (RU) наук. — М., 2007, с. 45-47, 77-84, 94-95 Изобретение относится к области биотехнологии, медицины, фармакологии, в частности к производству селективного активатора Са 2+-зависимой NO-синтазы (далее NOS), которая является одним из ключевых ферментов антиоксидантной защиты организма, а также эффективным регулятором Сазависимых процессов в стволовых клетках, что особенно важно для управления функциональными механизмами стволовой клетки in vivo. Известно действие динитрозильных комплексов железа (ДНКЖ) в качестве активатора Са 2+зависимой NOS [Vanin А.Р., Stukan R.A., Manukhina Е.В. Physical properties of dinitrosyl iron complexes inrelation with their vasodilator activity, Biochim Biophys Acta, 1996, 1295: 5-12]. Известно также действие иономицина как активатора Са 2+-зависимой NOS [Orlova E.V., UvarovV.W., Astashkin E.I. Effect of ionomycin on catalitic activity of nitric oxide synthase (NOS) from from rat thymocytes// In: Materials of the XIth International symposium of microsoms and drug oxidations. Los-Angeles,USA, 1996, p. 186; Орлова Е.В. Са 2+-зависимая NOS из тимоцитов и гепатоцитов крыс Wistar в онтогенезе.// Вопросы биологической, медицинской и фармацевтической химии, 2005, т. 4, с. 21-25]. Известно действие Т-активина как активатора Са 2+-зависимой NOS [Орлова Е.В. Влияние Са 2+антагониста верапамила и двух иммуномодуляторов: Т-активина и циклофосфамида на Са-зависимуюNOS из тимоцитов и гепатоцитов крыс Wistar// Вопросы биологической, медицинской и фармацевтической химии, 2005, т.
Известно действие динитрозильных комплексов железа (ДНКЖ) в качестве активатора Са 2+зависимой NOS [Vanin А.Р., Stukan R.A., Manukhina Е.В. Physical properties of dinitrosyl iron complexes inrelation with their vasodilator activity, Biochim Biophys Acta, 1996, 1295: 5-12]. Известно также действие иономицина как активатора Са 2+-зависимой NOS [Orlova E.V., UvarovV.W., Astashkin E.I. Effect of ionomycin on catalitic activity of nitric oxide synthase (NOS) from from rat thymocytes// In: Materials of the XIth International symposium of microsoms and drug oxidations. Los-Angeles,USA, 1996, p. 186; Орлова Е.В. Са 2+-зависимая NOS из тимоцитов и гепатоцитов крыс Wistar в онтогенезе.// Вопросы биологической, медицинской и фармацевтической химии, 2005, т. 4, с. 21-25]. Известно действие Т-активина как активатора Са 2+-зависимой NOS [Орлова Е.В. Влияние Са 2+антагониста верапамила и двух иммуномодуляторов: Т-активина и циклофосфамида на Са-зависимуюNOS из тимоцитов и гепатоцитов крыс Wistar// Вопросы биологической, медицинской и фармацевтической химии, 2005, т. 4, с. 26-29]. Недостатком этих препаратов является низкая степень активации Са 2+-зависимой NOS, а также сложная методика получения препаратов и высокие требования, предъявляемые к качеству исходного сырья. Техническим результатом, на получение которого направлено изобретение, является разработка селективного активатора Са 2+-зависимой NOS, характеризующегося высокой степенью активации, а также простого способа его изготовления, отличающегося простотой изготовления и доступностью необходимого для этого сырья. Технический результат достигается в селективном активаторе Са 2+-зависимой NOS, полученном следующим образом: дрожжи Saccharomyces cerevisiae (пекарские дрожжи), выращенные на синтетической среде с глюкозой, суспендируют в водопроводной воде, нагретой до 60-75 С, до концентрации 50 г сухой биомассы на 1 л, нагревают до 75-90 С и выдерживают в течение 15-25 мин при этой температуре,затем охлаждают до 35-60 С и инкубируют при этой температуре в течение 1,5-2,5 ч. Затем рН суспензии доводят до 1,0-1,5, снова нагревают до 75-80 С и выдерживают в течение 30-60 мин.
4, с. 26-29]. Недостатком этих препаратов является низкая степень активации Са 2+-зависимой NOS, а также сложная методика получения препаратов и высокие требования, предъявляемые к качеству исходного сырья. Техническим результатом, на получение которого направлено изобретение, является разработка селективного активатора Са 2+-зависимой NOS, характеризующегося высокой степенью активации, а также простого способа его изготовления, отличающегося простотой изготовления и доступностью необходимого для этого сырья. Технический результат достигается в селективном активаторе Са 2+-зависимой NOS, полученном следующим образом: дрожжи Saccharomyces cerevisiae (пекарские дрожжи), выращенные на синтетической среде с глюкозой, суспендируют в водопроводной воде, нагретой до 60-75 С, до концентрации 50 г сухой биомассы на 1 л, нагревают до 75-90 С и выдерживают в течение 15-25 мин при этой температуре,затем охлаждают до 35-60 С и инкубируют при этой температуре в течение 1,5-2,5 ч. Затем рН суспензии доводят до 1,0-1,5, снова нагревают до 75-80 С и выдерживают в течение 30-60 мин. Затем суспензию разводят водой с температурой от 2 до 25 С в соотношении от 1:1 до 1:10 и после разведения доводят рН до 5.0-6.0 и выдерживают при помешивании 30-60 мин. Далее надосадочную жидкость центрифугированием отделяют от клеточной биомассы дрожжей и затем высушивают. На фиг. 1 показан всплеск Са в клетках с 2 с 12 в ответ на добавление ДНКЖ (1.8 мкМ) — А и селективного активатора (0.005 мг/мл), по оси х отложено время t в минутах, а по оси у — относительные единицы флюоресценции. На фиг. 2 показана флуоресценция (0.005% Pluronic) в клетках с 2 с 12 в ответ на добавление ДНКЖ(1.8 мкМ) — А и селективного активатора (0.005 мг/мл) — В на лазерном сканирующем конфокальном микроскопе. Пример 1. Получение селективного активатора Са 2+-зависимой NOS. Дрожжи Saccharomyces cerevisiae, выращенные на синтетической среде с глюкозой, суспендируют в водопроводной воде, нагретой до 75 С до концентрации 50 г сухой биомассы на 1 л, нагревают до 75 С и выдерживают 20 мин, затем охлаждают до 35 С и инкубируют при этой температуре в течение 2 ч.
Затем суспензию разводят водой с температурой от 2 до 25 С в соотношении от 1:1 до 1:10 и после разведения доводят рН до 5.0-6.0 и выдерживают при помешивании 30-60 мин. Далее надосадочную жидкость центрифугированием отделяют от клеточной биомассы дрожжей и затем высушивают. На фиг. 1 показан всплеск Са в клетках с 2 с 12 в ответ на добавление ДНКЖ (1.8 мкМ) — А и селективного активатора (0.005 мг/мл), по оси х отложено время t в минутах, а по оси у — относительные единицы флюоресценции. На фиг. 2 показана флуоресценция (0.005% Pluronic) в клетках с 2 с 12 в ответ на добавление ДНКЖ(1.8 мкМ) — А и селективного активатора (0.005 мг/мл) — В на лазерном сканирующем конфокальном микроскопе. Пример 1. Получение селективного активатора Са 2+-зависимой NOS. Дрожжи Saccharomyces cerevisiae, выращенные на синтетической среде с глюкозой, суспендируют в водопроводной воде, нагретой до 75 С до концентрации 50 г сухой биомассы на 1 л, нагревают до 75 С и выдерживают 20 мин, затем охлаждают до 35 С и инкубируют при этой температуре в течение 2 ч.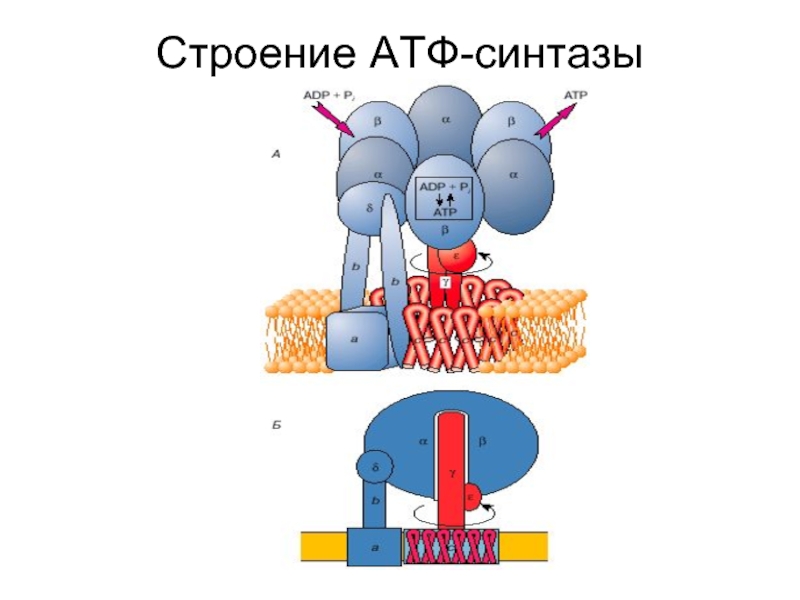 Затем рН суспензии доводят до 1.0 концентрированной h3SO4 и снова нагревают до 80 С в течение 60 мин. Затем разводят суспензию водой температурой 15 С в соотношении 1:5 и повышают рН среды до 5.8 концентрированной KOH. После разведения суспензию выдерживают 30-60 мин при помешивании и далее надосадочную жидкость центрифугированием отделяют от клеточной биомассы дрожжей и затем высушивают. Таблица 1 Влияние показателя разведения в способе перед отделением центрифугированием на активность Са 2+-зависимой NOS Пример 2. В табл. 2 приведены результаты исследований влияния селективного активатора Са 2+-зависимойNOS, полученного из дрожжей Sacchromyces cerevisiae раса 14, способом, описанным в примере 1, иономицина и Т-активина при 3-кратном в/б введении крысам Wistar на кинетические параметры Са 2+зависимой NOS. Таблица 2 По выраженности активирующего действия (уменьшение Km и увеличение удельной скорости реакции (Vуд селективный активатор Са 2+-зависимой NOS в использованной дозе 10 мг/кг является более сильным активатором Са 2+-зависимой NOS по сравнению с коммерческим пептидным препартом Тактивином и иономицином.
Затем рН суспензии доводят до 1.0 концентрированной h3SO4 и снова нагревают до 80 С в течение 60 мин. Затем разводят суспензию водой температурой 15 С в соотношении 1:5 и повышают рН среды до 5.8 концентрированной KOH. После разведения суспензию выдерживают 30-60 мин при помешивании и далее надосадочную жидкость центрифугированием отделяют от клеточной биомассы дрожжей и затем высушивают. Таблица 1 Влияние показателя разведения в способе перед отделением центрифугированием на активность Са 2+-зависимой NOS Пример 2. В табл. 2 приведены результаты исследований влияния селективного активатора Са 2+-зависимойNOS, полученного из дрожжей Sacchromyces cerevisiae раса 14, способом, описанным в примере 1, иономицина и Т-активина при 3-кратном в/б введении крысам Wistar на кинетические параметры Са 2+зависимой NOS. Таблица 2 По выраженности активирующего действия (уменьшение Km и увеличение удельной скорости реакции (Vуд селективный активатор Са 2+-зависимой NOS в использованной дозе 10 мг/кг является более сильным активатором Са 2+-зависимой NOS по сравнению с коммерческим пептидным препартом Тактивином и иономицином. Пример 3. Для изучения потенциальной способности селективного активатора Са 2+-зависимой NOS оказывать влияние на функциональные особенности стволовых клеток были проведены нижеследующие эксперименты. Культуру мышиных миобластов (как ближайшую к человеку по геномным характеристикамNOS в недифференцированных с 2 с 12 методом ЭПР-спектроскопии было проведено по методу Lancaster и Hibbs (Lancaster J.R., Hibbs J.B. EPR demonstration of iron-nitrosyl complex formation by cytotoxic activated macrophages// Proc. Natl. Acad. Sci. USA, v. 87, p. 1223-1227, 1999). Результаты приведены в табл. 3. Таблица 3 Изучение Са-сигнализации в присутствии флюоресцентного красителя 0.005% Pluronic проводили по методу, описанному в работе Gutierrez-Martin Y. и Martin-Romero F.J. (Gutierrez-Martin Y., MartinRomero F.J., Henao F. Store-operated calcium entry in differentiated C2C12 skeletal muscle cells// Biochim. etbiophys. acta., 2005, v. 1711, p. 33-40). Подсчет клеток проводили в камере Горяева. На фиг. 1 и 2 видно,что селективный активатор Са 2+-зависимой NOS (0.
Пример 3. Для изучения потенциальной способности селективного активатора Са 2+-зависимой NOS оказывать влияние на функциональные особенности стволовых клеток были проведены нижеследующие эксперименты. Культуру мышиных миобластов (как ближайшую к человеку по геномным характеристикамNOS в недифференцированных с 2 с 12 методом ЭПР-спектроскопии было проведено по методу Lancaster и Hibbs (Lancaster J.R., Hibbs J.B. EPR demonstration of iron-nitrosyl complex formation by cytotoxic activated macrophages// Proc. Natl. Acad. Sci. USA, v. 87, p. 1223-1227, 1999). Результаты приведены в табл. 3. Таблица 3 Изучение Са-сигнализации в присутствии флюоресцентного красителя 0.005% Pluronic проводили по методу, описанному в работе Gutierrez-Martin Y. и Martin-Romero F.J. (Gutierrez-Martin Y., MartinRomero F.J., Henao F. Store-operated calcium entry in differentiated C2C12 skeletal muscle cells// Biochim. etbiophys. acta., 2005, v. 1711, p. 33-40). Подсчет клеток проводили в камере Горяева. На фиг. 1 и 2 видно,что селективный активатор Са 2+-зависимой NOS (0. 005 мг/мл) сильнее активирует Са-каналы, чем ДНКЖ в концентрации 1.8 мкМ. Таким образом, можно заключить, что селективный активатор Са 2+зависимой NOS активирует NO-синтазу в недифференцированных с 2 с 12 в гораздо большей степени, чем динитрозийный комплекс железа и иономицин. При этом, как следует из экспериментов по Сасигнализации, в недифференцированных с 2 с 12 активируется именно Са 2+-зависимая NOS, а также эффективно увеличивает выход Са из клеточных депо в цитоплазму. Функцию «депо» NO в клетке, наряду с нитрозотиолами, как известно, выполняют нитрозильные негемовые [Fe-S] белки. Синтетические аналоги динитрозильных комплексов железа с природными тиолами (цистеином, глютатионом и др.) (ДНКЖ) представляют собой малостабильные водные растворы и идентифицируются по ЭПР сигналу с гиромагнитным отношением g=2.03 (Vanin A.F., Stukan R.A., Manukhina E.B. Physical properties of dinitrosyl iron complexes in relation with their vasodilator activity//Biochim. Biophys. Acta, 1996, v. 1295, p.
005 мг/мл) сильнее активирует Са-каналы, чем ДНКЖ в концентрации 1.8 мкМ. Таким образом, можно заключить, что селективный активатор Са 2+зависимой NOS активирует NO-синтазу в недифференцированных с 2 с 12 в гораздо большей степени, чем динитрозийный комплекс железа и иономицин. При этом, как следует из экспериментов по Сасигнализации, в недифференцированных с 2 с 12 активируется именно Са 2+-зависимая NOS, а также эффективно увеличивает выход Са из клеточных депо в цитоплазму. Функцию «депо» NO в клетке, наряду с нитрозотиолами, как известно, выполняют нитрозильные негемовые [Fe-S] белки. Синтетические аналоги динитрозильных комплексов железа с природными тиолами (цистеином, глютатионом и др.) (ДНКЖ) представляют собой малостабильные водные растворы и идентифицируются по ЭПР сигналу с гиромагнитным отношением g=2.03 (Vanin A.F., Stukan R.A., Manukhina E.B. Physical properties of dinitrosyl iron complexes in relation with their vasodilator activity//Biochim. Biophys. Acta, 1996, v. 1295, p.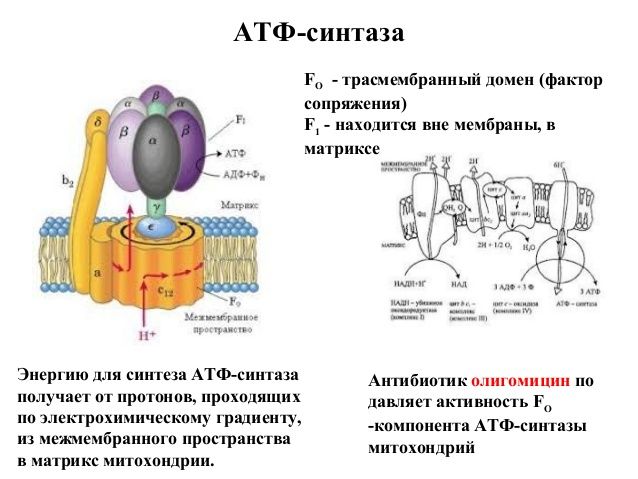 5-12). Таким образом, достигается технический результат. Кроме того, селективный активатор Са 2+зависимой NOS отличается пониженным содержанием нуклеиновых кислот, что снижает вероятность возникновения мочекаменной болезни при его применении. ФОРМУЛА ИЗОБРЕТЕНИЯ Способ получения селективного активатора Са 2+-зависимой NO-синтазы, характеризующийся тем,что дрожжи Saccharomyces cerevisiae, выращенные на синтетической среде с глюкозой, суспендируют в водопроводной воде, нагретой до 60-75 С, до концентрации 50 г сухой биомассы на 1 л, нагревают до 7590 С и выдерживают в течение 15-25 мин при этой температуре, затем охлаждают до 35-60 С, инкубируют при этой температуре в течение 1,5-2,5 ч, после чего рН суспензии доводят до 1,0-1,5, повторно нагревают до 75-85 С и выдерживают в течение 30-60 мин, затем суспензию разводят водой в соотношении от 1:1 до 1:10 и доводят рН суспензии до 5.0-6.0, выдерживают при помешивании 30-60 мин, после чего надосадочную жидкость центрифугированием отделяют от клеточной биомассы дрожжей и затем высушивают.
5-12). Таким образом, достигается технический результат. Кроме того, селективный активатор Са 2+зависимой NOS отличается пониженным содержанием нуклеиновых кислот, что снижает вероятность возникновения мочекаменной болезни при его применении. ФОРМУЛА ИЗОБРЕТЕНИЯ Способ получения селективного активатора Са 2+-зависимой NO-синтазы, характеризующийся тем,что дрожжи Saccharomyces cerevisiae, выращенные на синтетической среде с глюкозой, суспендируют в водопроводной воде, нагретой до 60-75 С, до концентрации 50 г сухой биомассы на 1 л, нагревают до 7590 С и выдерживают в течение 15-25 мин при этой температуре, затем охлаждают до 35-60 С, инкубируют при этой температуре в течение 1,5-2,5 ч, после чего рН суспензии доводят до 1,0-1,5, повторно нагревают до 75-85 С и выдерживают в течение 30-60 мин, затем суспензию разводят водой в соотношении от 1:1 до 1:10 и доводят рН суспензии до 5.0-6.0, выдерживают при помешивании 30-60 мин, после чего надосадочную жидкость центрифугированием отделяют от клеточной биомассы дрожжей и затем высушивают.
<a href=»https://easpatents.com/4-24447-sposob-polucheniya-selektivnogo-aktivatora-ca2-zavisimojj-no-sintazy.html» rel=»bookmark» title=»База патентов Евразийского Союза»>Способ получения селективного активатора ca2+-зависимой no-синтазы</a>
Синтазы оксида азота: регуляция и функция
Eur Heart J. 2012 Apr; 33 (7): 829–837.
Ульрих Фёрстерманн
1 Кафедра фармакологии, Медицинский центр Университета Йоханнеса Гутенберга, 55101 Майнц, Германия
Уильям С. Сесса
2 Кафедра фармакологии, биологии сосудов и терапевтической программы Медицинской школы Йельского университета , New Haven CT 06520, USA
1 Кафедра фармакологии, Медицинский центр Университета Йоханнеса Гутенберга, 55101 Майнц, Германия
2 Кафедра фармакологии и биологии сосудов и программа терапии Медицинской школы Йельского университета, Нью-Хейвен CT 06520, USA
* Автор, ответственный за переписку: Отдел фармакологии, Медицинский центр Университета Йоханнеса Гутенберга, Obere Zahlbacher Strasse 67, 55131 Mainz, Германия. Тел .: +49 6131 17 9150, Факс: +49 6131 17 9043, Электронная почта: [email protected]
Тел .: +49 6131 17 9150, Факс: +49 6131 17 9043, Электронная почта: [email protected]Получено 7 января 2011 г .; Пересмотрено 14 июля 2011 г .; Принято 28 июля 2011 г.
Авторские права Опубликовано от имени Европейского общества кардиологов. Все права защищены. © Автор 2011. Для получения разрешения, пожалуйста, напишите: [email protected] Эта статья цитируется другими статьями в PMC.Abstract
Оксид азота (NO), самая маленькая из известных сигнальных молекул, продуцируется тремя изоформами NO-синтазы (NOS; EC 1.14.13.39). Все они используют l-аргинин и молекулярный кислород в качестве субстратов и требуют, чтобы кофакторы восстановили никотинамид-аденин-динуклеотидфосфат (НАДФН), флавинадениндинуклеотид (FAD), флавинмононуклеотид (FMN) и (6 R -) 5,6 , 7,8-тетрагидробиоптерин (BH 4 ). Все БДУ связывают кальмодулин и содержат гем. Нейрональная NOS (nNOS, NOS I) конститутивно экспрессируется в центральных и периферических нейронах и некоторых других типах клеток. Его функции включают синаптическую пластичность в центральной нервной системе (ЦНС), центральную регуляцию артериального давления, расслабление гладких мышц и расширение сосудов через периферические нитрергические нервы.Нитрергические нервы имеют особое значение для расслабления кавернозного тела и эрекции полового члена. Ингибиторы фосфодиэстеразы 5 (силденафил, варденафил и тадалафил) требуют, по крайней мере, остаточной активности nNOS для их действия. Индуцибельная NOS (NOS II) может экспрессироваться во многих типах клеток в ответ на липополисахарид, цитокины или другие агенты. Индуцибельная NOS генерирует большие количества NO, которые оказывают цитостатическое действие на паразитические клетки-мишени. Индуцируемая БДУ способствует патофизиологии воспалительных заболеваний и септического шока.Эндотелиальная NOS (eNOS, NOS III) в основном экспрессируется в эндотелиальных клетках. Он поддерживает расширение кровеносных сосудов, контролирует кровяное давление и обладает множеством других вазопротекторных и антиатеросклеротических эффектов.
Его функции включают синаптическую пластичность в центральной нервной системе (ЦНС), центральную регуляцию артериального давления, расслабление гладких мышц и расширение сосудов через периферические нитрергические нервы.Нитрергические нервы имеют особое значение для расслабления кавернозного тела и эрекции полового члена. Ингибиторы фосфодиэстеразы 5 (силденафил, варденафил и тадалафил) требуют, по крайней мере, остаточной активности nNOS для их действия. Индуцибельная NOS (NOS II) может экспрессироваться во многих типах клеток в ответ на липополисахарид, цитокины или другие агенты. Индуцибельная NOS генерирует большие количества NO, которые оказывают цитостатическое действие на паразитические клетки-мишени. Индуцируемая БДУ способствует патофизиологии воспалительных заболеваний и септического шока.Эндотелиальная NOS (eNOS, NOS III) в основном экспрессируется в эндотелиальных клетках. Он поддерживает расширение кровеносных сосудов, контролирует кровяное давление и обладает множеством других вазопротекторных и антиатеросклеротических эффектов. Многие факторы риска сердечно-сосудистых заболеваний приводят к окислительному стрессу, разъединению eNOS и дисфункции эндотелия в сосудистой сети. Фармакологически оксидативный стресс сосудов можно уменьшить и восстановить функциональность eNOS с помощью ингибиторов ренин- и ангиотензинпревращающего фермента, блокаторов рецепторов ангиотензина и статинов.
Многие факторы риска сердечно-сосудистых заболеваний приводят к окислительному стрессу, разъединению eNOS и дисфункции эндотелия в сосудистой сети. Фармакологически оксидативный стресс сосудов можно уменьшить и восстановить функциональность eNOS с помощью ингибиторов ренин- и ангиотензинпревращающего фермента, блокаторов рецепторов ангиотензина и статинов.
Ключевые слова: (6 R -) 5,6,7,8-тетрагидробиоптерин; l-аргинин; Асимметричный диметил-1-аргинин; НАДФН-оксидаза; Пероксинитрит
Введение
Оксид азота (NO) — неортодоксальная молекула-посредник, которая имеет множество молекулярных мишеней. NO контролирует серворегуляторные функции, такие как нейротрансмиссия 1,2 или тонус сосудов 3,4 (путем стимуляции NO-чувствительной гуанилилциклазы), регулирует транскрипцию генов 5,6 и трансляцию мРНК (например,грамм. путем связывания с элементами, чувствительными к железу), 7,8 и производит посттрансляционные модификации белков (например, путем рибозилирования АДФ). 9,10 Важным способом инактивации NO является его реакция с супероксид-анионом (O 2 — • ) с образованием мощного окислителя пероксинитрита (ONOO — ). Это соединение может вызывать окислительное повреждение, нитрование и S-нитрозилирование биомолекул, включая белки, липиды и ДНК. 11,12 Нитрозативный стресс, вызванный ONOO — , был вовлечен в разрыв однонитевой ДНК с последующей активацией поли-АДФ-рибозо-полимеразы (PARP). 13
9,10 Важным способом инактивации NO является его реакция с супероксид-анионом (O 2 — • ) с образованием мощного окислителя пероксинитрита (ONOO — ). Это соединение может вызывать окислительное повреждение, нитрование и S-нитрозилирование биомолекул, включая белки, липиды и ДНК. 11,12 Нитрозативный стресс, вызванный ONOO — , был вовлечен в разрыв однонитевой ДНК с последующей активацией поли-АДФ-рибозо-полимеразы (PARP). 13
У млекопитающих NO может генерироваться тремя различными изоформами фермента NO-синтазы (NOS; l-аргинин, NADPH: оксидоредуктазы кислорода, образование NO; EC 1.14.13.39). Изоферменты называются нейронными «n’NOS» (или NOS I), индуцибельными «i’NOS» (или NOS II) и эндотелиальными «e’NOS» (или NOS III) ( фиг. ). В этом обзоре будут рассмотрены все три изоформы. Однако основное внимание в статье будет уделено eNOS и функциям NO в сердечно-сосудистой системе.
Важные функции различных изоформ NOS. (Верхняя панель) NOS нейронов экспрессируется в определенных нейронах центральной нервной системы (ЦНС). Это было связано с синаптической пластичностью (то есть такими явлениями, как долгосрочное потенцирование и долгосрочное торможение). Эти явления участвуют в обучении и формировании памяти. NO, полученный из нейронов NOS, также участвует в центральном контроле артериального давления. В периферической нервной системе (ПНС) нейрональный NO, полученный из NOS, действует как атипичный нейромедиатор, который опосредует расслабляющие компоненты перистальтики кишечника, вазодилатацию и эрекцию полового члена.По крайней мере, минимальная стимуляция растворимой гуанилилциклазы в пещеристом теле NO, производным от nNOS, и последующее образование небольших количеств циклического GMP является предпосылкой для проэректильного действия ингибиторов фосфодиэстеразы 5 силденафила (Виагра ® ). , варденафил (Левитра ® ) и тадалафил (Сиалис ® ). (Средняя панель) Индуцируемая экспрессия NOS может быть индуцирована цитокинами и другими агентами практически в любом типе клеток.
(Верхняя панель) NOS нейронов экспрессируется в определенных нейронах центральной нервной системы (ЦНС). Это было связано с синаптической пластичностью (то есть такими явлениями, как долгосрочное потенцирование и долгосрочное торможение). Эти явления участвуют в обучении и формировании памяти. NO, полученный из нейронов NOS, также участвует в центральном контроле артериального давления. В периферической нервной системе (ПНС) нейрональный NO, полученный из NOS, действует как атипичный нейромедиатор, который опосредует расслабляющие компоненты перистальтики кишечника, вазодилатацию и эрекцию полового члена.По крайней мере, минимальная стимуляция растворимой гуанилилциклазы в пещеристом теле NO, производным от nNOS, и последующее образование небольших количеств циклического GMP является предпосылкой для проэректильного действия ингибиторов фосфодиэстеразы 5 силденафила (Виагра ® ). , варденафил (Левитра ® ) и тадалафил (Сиалис ® ). (Средняя панель) Индуцируемая экспрессия NOS может быть индуцирована цитокинами и другими агентами практически в любом типе клеток. Первоначально это было показано для макрофагов (MΦ).Индукция индуцибельной NOS в MΦ необходима для борьбы с внутриклеточными бактериями, такими как Mycobacterium tuberculosis 156,157 или паразитом Leishmania . 158,159 Однако индуцибельная NOS также активируется при различных типах воспалительных заболеваний, и NO, генерируемый ферментом, опосредует различные симптомы воспаления. 160,161 Наконец, индуцибельный NO, производный от NOS, является преобладающим медиатором вазодилатации и падения артериального давления, наблюдаемого при септическом шоке. 161 Фактически, мыши с нарушенным индуцибельным геном NOS защищены от многих симптомов септического шока. 159 (Нижняя панель) NO, полученный из эндотелиальной NOS, является физиологическим вазодилататором, но также может обеспечивать вазопротекцию несколькими способами. NO, выделяемый в просвет сосудов, является мощным ингибитором агрегации тромбоцитов и их адгезии к стенке сосудов.
Первоначально это было показано для макрофагов (MΦ).Индукция индуцибельной NOS в MΦ необходима для борьбы с внутриклеточными бактериями, такими как Mycobacterium tuberculosis 156,157 или паразитом Leishmania . 158,159 Однако индуцибельная NOS также активируется при различных типах воспалительных заболеваний, и NO, генерируемый ферментом, опосредует различные симптомы воспаления. 160,161 Наконец, индуцибельный NO, производный от NOS, является преобладающим медиатором вазодилатации и падения артериального давления, наблюдаемого при септическом шоке. 161 Фактически, мыши с нарушенным индуцибельным геном NOS защищены от многих симптомов септического шока. 159 (Нижняя панель) NO, полученный из эндотелиальной NOS, является физиологическим вазодилататором, но также может обеспечивать вазопротекцию несколькими способами. NO, выделяемый в просвет сосудов, является мощным ингибитором агрегации тромбоцитов и их адгезии к стенке сосудов. Помимо защиты от тромбоза, это также предотвращает высвобождение тромбоцитарных факторов роста, которые стимулируют пролиферацию гладких мышц и производство матричных молекул.Эндотелиальный NO также контролирует экспрессию генов, участвующих в атерогенезе. NO снижает экспрессию хемоаттрактантного белка MCP-1 и ряда молекул поверхностной адгезии, тем самым предотвращая адгезию лейкоцитов к эндотелию сосудов и миграцию лейкоцитов в стенку сосудов. Это обеспечивает защиту от ранних фаз атерогенеза. Также сниженная проницаемость эндотелия, уменьшенный приток липопротеинов в сосудистую стенку и ингибирование окисления липопротеинов низкой плотности могут вносить вклад в антиатерогенные свойства NO, полученного из эндотелиальной NOS.Наконец, было показано, что NO ингибирует синтез ДНК, митогенез и пролиферацию гладкомышечных клеток сосудов, а также миграцию гладкомышечных клеток, тем самым защищая от более поздней фазы атерогенеза. Основываясь на комбинации этих эффектов, NO, продуцируемый в эндотелиальных клетках, можно рассматривать как антиатеросклеротический принцип (см.
Помимо защиты от тромбоза, это также предотвращает высвобождение тромбоцитарных факторов роста, которые стимулируют пролиферацию гладких мышц и производство матричных молекул.Эндотелиальный NO также контролирует экспрессию генов, участвующих в атерогенезе. NO снижает экспрессию хемоаттрактантного белка MCP-1 и ряда молекул поверхностной адгезии, тем самым предотвращая адгезию лейкоцитов к эндотелию сосудов и миграцию лейкоцитов в стенку сосудов. Это обеспечивает защиту от ранних фаз атерогенеза. Также сниженная проницаемость эндотелия, уменьшенный приток липопротеинов в сосудистую стенку и ингибирование окисления липопротеинов низкой плотности могут вносить вклад в антиатерогенные свойства NO, полученного из эндотелиальной NOS.Наконец, было показано, что NO ингибирует синтез ДНК, митогенез и пролиферацию гладкомышечных клеток сосудов, а также миграцию гладкомышечных клеток, тем самым защищая от более поздней фазы атерогенеза. Основываясь на комбинации этих эффектов, NO, продуцируемый в эндотелиальных клетках, можно рассматривать как антиатеросклеротический принцип (см. Обзор Li and Förstermann 162 ).
Обзор Li and Förstermann 162 ).
Механизмы синтеза оксида азота
Все изоформы NOS используют l-аргинин в качестве субстрата, а также молекулярный кислород и восстановленный никотинамид-аденин-динуклеотидфосфат (НАДФН) в качестве субстратов.Флавинадениндинуклеотид (FAD), флавинмононуклеотид (FMN) и (6 R -) 5,6,7,8-тетрагидро-1-биоптерин (BH 4 ) являются кофакторами всех изоферментов. Все белки NOS являются гомодимерами ( фиг. ). Функциональная NOS переносит электроны от NADPH через флавины FAD и FMN в карбоксиконцевом редуктазном домене к гемму в аминоконцевом оксигеназном домене. Домен оксигеназы также связывает важный кофактор BH 4 , молекулярный кислород и субстрат l-аргинин 14,15 ( Рисунок ).На участке гема электроны используются для восстановления и активации O 2 и для окисления l-аргинина до l-цитруллина и NO ( Рисунок ). Последовательности, расположенные рядом с цистеиновым лигандом гема, также, по-видимому, участвуют в связывании l-аргинина и BH 4 . 16 Чтобы синтезировать NO, фермент NOS проходит два этапа. На первом этапе NOS гидроксилирует l-аргинин до N ω -гидрокси-1-аргинина (который остается в значительной степени связанным с ферментом).На втором этапе NOS окисляет N ω -гидрокси-1-аргинин до 1-цитруллина и NO. 17,18 Все изоформы NOS связывают кальмодулин ( фигура ). В nNOS и eNOS связывание кальмодулина вызывается увеличением внутриклеточного Ca 2+ (полумаксимальная активность между 200 и 400 нМ). Когда сродство кальмодулина к NOS увеличивается, это облегчает поток электронов от НАДФН в домене редуктазы к гему в домене оксигеназы. В индуцибельной NOS (iNOS) кальмодулин связывается уже при чрезвычайно низких внутриклеточных концентрациях Ca 2+ (ниже 40 нМ) из-за другой аминокислотной структуры сайта связывания кальмодулина. 19,20 Все белки NOS содержат цинк-тиолатный кластер, образованный ионом цинка, который тетраэдрически координирован с двумя мотивами CysXXXXCys (по одному от каждого мономера) на границе димера NOS.
16 Чтобы синтезировать NO, фермент NOS проходит два этапа. На первом этапе NOS гидроксилирует l-аргинин до N ω -гидрокси-1-аргинина (который остается в значительной степени связанным с ферментом).На втором этапе NOS окисляет N ω -гидрокси-1-аргинин до 1-цитруллина и NO. 17,18 Все изоформы NOS связывают кальмодулин ( фигура ). В nNOS и eNOS связывание кальмодулина вызывается увеличением внутриклеточного Ca 2+ (полумаксимальная активность между 200 и 400 нМ). Когда сродство кальмодулина к NOS увеличивается, это облегчает поток электронов от НАДФН в домене редуктазы к гему в домене оксигеназы. В индуцибельной NOS (iNOS) кальмодулин связывается уже при чрезвычайно низких внутриклеточных концентрациях Ca 2+ (ниже 40 нМ) из-за другой аминокислотной структуры сайта связывания кальмодулина. 19,20 Все белки NOS содержат цинк-тиолатный кластер, образованный ионом цинка, который тетраэдрически координирован с двумя мотивами CysXXXXCys (по одному от каждого мономера) на границе димера NOS. 21–23 Цинк в БДУ выполняет структурную, а не каталитическую функцию. 21
21–23 Цинк в БДУ выполняет структурную, а не каталитическую функцию. 21
Структура и каталитические механизмы функциональных БДУ. ( A ) Мономеры NOS способны переносить электроны от восстановленного никотинамид-аденин-динуклеотидфосфата (NADPH) к флавина-аденин-динуклеотиду (FAD) и флавин-мононуклеотиду (FMN) и обладают ограниченной способностью восстанавливать молекулярный кислород до супероксид (O 2 — • ). 18,163,164 Мономеры и изолированные домены редуктазы могут связывать кальмодулин (CaM), что усиливает перенос электронов внутри домена редуктазы. 165 Мономеры NOS не способны связывать кофактор (6 R -) 5,6,7,8-тетрагидробиоптерин (BH 4 ) или субстрат l-аргинин и не могут катализировать продукцию NO. 163 166 ( B ) В присутствии гема NOS может образовывать функциональный димер. 163,166 Гем необходим для междоменного переноса электронов от флавинов к гему противоположного мономера. 165,167 Из-за различий в кальмодулин-связывающем домене, повышенный Ca 2+ необходим для связывания кальмодулина (и, следовательно, каталитической активности) в nNOS и eNOS, тогда как кальмодулин связывается с индуцибельными NOS с высоким сродством даже в отсутствие Ca 2+ . Когда присутствует достаточное количество субстрата l-аргинина (l-Arg) и кофактора BH 4 , интактные димеры NOS связывают свой гем и восстановление O 2 с синтезом NO (полностью функциональный NOS). l-цитруллин (l-Cit) образуется как побочный продукт.Для ясности показан только поток электронов от редуктазного домена одного мономера к оксигеназному домену другого мономера. Ферменты NOS выполняют две отдельные стадии окисления: одну для образования N ω -гидрокси-1-аргинина, а вторую — для превращения этого промежуточного продукта в NO. 18 Все изоформы NOS содержат ион цинка (Zn), координированный в тетраэдрической конформации с парами мотивов CXXXXC на границе раздела димеров.
165,167 Из-за различий в кальмодулин-связывающем домене, повышенный Ca 2+ необходим для связывания кальмодулина (и, следовательно, каталитической активности) в nNOS и eNOS, тогда как кальмодулин связывается с индуцибельными NOS с высоким сродством даже в отсутствие Ca 2+ . Когда присутствует достаточное количество субстрата l-аргинина (l-Arg) и кофактора BH 4 , интактные димеры NOS связывают свой гем и восстановление O 2 с синтезом NO (полностью функциональный NOS). l-цитруллин (l-Cit) образуется как побочный продукт.Для ясности показан только поток электронов от редуктазного домена одного мономера к оксигеназному домену другого мономера. Ферменты NOS выполняют две отдельные стадии окисления: одну для образования N ω -гидрокси-1-аргинина, а вторую — для превращения этого промежуточного продукта в NO. 18 Все изоформы NOS содержат ион цинка (Zn), координированный в тетраэдрической конформации с парами мотивов CXXXXC на границе раздела димеров. Этот сайт имеет большое значение для связывания BH 4 и l-аргинина.Перенос электрона из домена редуктазы (*) позволяет NOS-феррику (Fe 3+ ) гему связывать O 2 и образовывать двухвалентный (Fe 2+ ) -диокси. Этот вид может получать второй электрон (**) предпочтительно от BH 4 или от домена редуктазы. Природа образовавшегося окисленного BH 4 была идентифицирована как радикал тригидробиоптерина (BH 3 • ) или катион-радикал тригидроптерина, протонированный по N5 (BH 3 • H + ).Радикал BH 3 • (или катион-радикал) может быть рециркулирован в BH 4 самой NOS (с использованием электрона, поставляемого флавинами). Альтернативно, есть доказательства того, что восстановители, такие как аскорбиновая кислота (AscH, который присутствует в клетках в миллимолярных концентрациях), могут восстанавливать радикал BH 3 • обратно до BH 4 103 (Asc · , аскорбат радикал).
Этот сайт имеет большое значение для связывания BH 4 и l-аргинина.Перенос электрона из домена редуктазы (*) позволяет NOS-феррику (Fe 3+ ) гему связывать O 2 и образовывать двухвалентный (Fe 2+ ) -диокси. Этот вид может получать второй электрон (**) предпочтительно от BH 4 или от домена редуктазы. Природа образовавшегося окисленного BH 4 была идентифицирована как радикал тригидробиоптерина (BH 3 • ) или катион-радикал тригидроптерина, протонированный по N5 (BH 3 • H + ).Радикал BH 3 • (или катион-радикал) может быть рециркулирован в BH 4 самой NOS (с использованием электрона, поставляемого флавинами). Альтернативно, есть доказательства того, что восстановители, такие как аскорбиновая кислота (AscH, который присутствует в клетках в миллимолярных концентрациях), могут восстанавливать радикал BH 3 • обратно до BH 4 103 (Asc · , аскорбат радикал).
NO, образованный NOS, может действовать на ряд целевых ферментов и белков.Наиболее важным физиологическим сигнальным путем, стимулируемым NO, является активация растворимой гуанилилциклазы и образование циклического GMP. 3,4,24–26
Нейрональная синтаза оксида азота
Нейрональная БДУ постоянно экспрессируется в определенных нейронах головного мозга ( Рисунок ). Активность фермента регулируется Ca 2+ и кальмодулином. Мозговая nNOS находится в клетках в виде частиц и растворимой формы, и дифференциальная субклеточная локализация nNOS может способствовать ее разнообразным функциям.NOS нейронов содержит домен PDZ и может напрямую взаимодействовать с доменами PDZ других белков. Эти взаимодействия определяют субклеточное распределение и активность фермента. 27 Помимо ткани головного мозга, nNOS была идентифицирована иммуногистохимическим методом в спинном мозге, в симпатических ганглиях и надпочечниках, в периферических нитрергических нервах, в эпителиальных клетках различных органов, в клетках макулы почки, в клетках островков поджелудочной железы. , и в гладких мышцах сосудов. 28 У млекопитающих крупнейшим источником nNOS с точки зрения массы ткани являются скелетные мышцы. 28,29
Физиологические функции нейрональной синтазы оксида азота
В последние годы все больше сообщений подтверждают важность nNOS в различных синаптических сигнальных событиях. Нейрональная БДУ участвует в модуляции физиологических функций, таких как обучение, память и нейрогенез. 27 В центральной нервной системе (ЦНС) nNOS опосредует долгосрочную регуляцию синаптической передачи (долгосрочное потенцирование, долгосрочное ингибирование), 1,2,30,31 , тогда как нет никаких доказательств вовлечения NO, происходящего от nNOS, при острой нейротрансмиссии (фиг. , фиг. ).Предполагается, что ретроградная коммуникация через синаптические соединения участвует в формировании памяти, и есть доказательства того, что ингибиторы NOS ухудшают обучение и вызывают амнезию в моделях на животных. 32,33 Есть также свидетельства того, что NO, образованный в ЦНС nNOS, участвует в центральной регуляции артериального давления 34–36 ( Рисунок ). Блокада активности nNOS в мозговом веществе и гипоталамусе вызывает системную гипертензию. 37
На периферии многие гладкомышечные ткани иннервируются нитрергическими нервами , i.е. нервы, которые содержат nNOS и генерируют и высвобождают NO (фиг. , ). Оксид азота, продуцируемый nNOS в нитрергических нервах, можно рассматривать как необычный нейромедиатор, который стимулирует NO-чувствительную гуанилилциклазу в ее эффекторных клетках, тем самым снижая тонус различных типов гладких мышц, включая кровеносные сосуды. 28,38 Общепринятое представление о том, что eNOS в основном отвечает за регуляцию сосудистого тонуса на периферии (см. Далее в этой статье), было оспорено в исследовании на людях с S -метил-1-тиоцитруллин (SMTC), селективный ингибитор nNOS. S -Метил-1-тиоцитруллин снижает базальный кровоток в предплечье человека и в коронарном кровообращении. Этот эффект можно обратить вспять с помощью l-аргинина. Интересно, что SMTC не влияет на классическую eNOS-опосредованную вазодилатацию в ответ на ацетилхолин, вещество P или напряжение сдвига жидкости. Эти данные согласуются с представлением о том, что nNOS играет важную роль в регуляции сосудистого тонуса, независимо от эффектов nNOS в ЦНС. Таким образом, eNOS и nNOS могут играть разные роли в физиологической регуляции тонуса микрососудов человека in vivo . 39 Интересно, что клетки гладких мышц сосудов также экспрессируют низкие уровни nNOS, которые, как было показано, поддерживают некоторую степень вазодилатации, когда преобладающая eNOS становится дисфункциональной. 40
Опосредуя расслабление гладких мышц кавернозного тела, нитрергические нервы, содержащие nNOS, ответственны за эрекцию полового члена 41,42 ( Рисунок ). Также в кавернозном теле NO-индуцированная релаксация гладких мышц опосредуется циклическим GMP. 42 Циклический GMP разлагается фосфодиэстеразами. Преобладающей изоформой в кавернозном теле является изоформа 5. 43 Таким образом, остаточная активность nNOS важна для проэректильного эффекта селективных ингибиторов фосфодиэстеразы 5, таких как силденафил (Виагра ® ), варденафил (Левитра ) и тадалафил (Сиалис ® ). 43,44 Интересно, что поскольку фосфодиэстераза 5 также значительно экспрессируется в легочных артериях, силденафил (под торговым названием Revatio ® ) и тадалафил (под торговым названием Adcirca ® ) также одобрен для лечения легочных артерий. артериальная гипертензия.
Роль нейрональной синтазы оксида азота в патофизиологии
Аномальная передача сигналов NO может вносить вклад в различные нейродегенеративные патологии, такие как эксайтотоксичность после инсульта, рассеянный склероз, болезни Альцгеймера и Паркинсона. 45 Гиперактивный nNOS, стимулированный массивным притоком Ca 2+ в нейрональные клетки, был вовлечен в опосредованную N -метил-d-аспартат рецептором гибель нейронов при цереброваскулярном инсульте. 46 В этих условиях NO может вносить вклад в эксайтотоксичность, вероятно, за счет пероксинитритной активации PARP и / или перехода митохондриальной проницаемости.Высокие уровни NO также могут вызывать истощение энергии из-за ингибирования митохондриального дыхания и ингибирования гликолиза. 47
Некоторые нарушения тонуса гладкой мускулатуры желудочно-кишечного тракта (например, гастроэзофагеальная рефлюксная болезнь) также могут быть связаны с гиперпродукцией NO посредством nNOS в периферических нитрергических нервах. 48,49
Индуцибельная синтаза оксида азота
Индуцибельная NOS обычно не экспрессируется в клетках, но ее экспрессия может быть индуцирована бактериальным липополисахаридом, цитокинами и другими агентами.Хотя в первую очередь идентифицируется в макрофагах ( фиг. ), экспрессия фермента может быть стимулирована практически в любой клетке или ткани при условии, что были идентифицированы соответствующие индуцирующие агенты. 28,38 После экспрессии iNOS постоянно активен и не регулируется внутриклеточными концентрациями Ca 2+ .
Физиологические функции индуцибельной синтазы оксида азота
Индуцибельная NOS, индуцируемая в макрофагах, производит большие количества NO, которые представляют собой основной цитотоксический принцип этих клеток. 50 Благодаря своему сродству к железу, связанному с белками, NO может ингибировать ключевые ферменты, которые содержат железо в своих каталитических центрах. К ним относятся железо-серные кластер-зависимые ферменты (комплексы I и II), участвующие в митохондриальном электронном транспорте, рибонуклеотидредуктаза (фермент, ограничивающий скорость репликации ДНК) и цис -конитаза (ключевой фермент в цикле лимонной кислоты). . 50 Кроме того, более высокие концентрации NO, продуцируемые индуцированными макрофагами, могут напрямую мешать ДНК клеток-мишеней и вызывать разрывы цепи и фрагментацию. 51,52 Сочетание этих эффектов, вероятно, лежит в основе цитостатического и цитотоксического действия NO на паразитические микроорганизмы и определенные опухолевые клетки ( фиг. ). Интересно, что неиммунные клетки также могут быть индуцированы цитокинами для высвобождения количества NO, достаточно большого, чтобы повлиять на соседние клетки. Например, было показано, что активированные цитокинами эндотелиальные клетки лизируют опухолевые клетки 53 , а индуцированные гепатоциты могут использовать NO для уничтожения спорозоитов малярии. 54 Индуцируемая активность NOS, вероятно, ответственна за все эти эффекты.
Роль индуцибельной синтазы оксида азота в патофизиологии. неправильный сайт — также может нанести вред здоровым клеткам.
In vivo повреждение клеток и тканей может быть связано с самим радикалом NO или взаимодействием NO с O 2 — • , приводящим к образованию пероксинитрита (ONOO — ).Подавляющее большинство воспалительных и аутоиммунных поражений характеризуется обилием активированных макрофагов и нейтрофилов. Эти клетки могут секретировать значительные количества NO, что приводит к повреждению окружающей ткани 52,55 ( Рисунок ). Индуцируемый NO, происходящий из NOS, также, вероятно, участвует в неспецифическом отторжении аллотрансплантата. 56Воспалительная нейродегенерация способствует развитию ряда патологий головного мозга. Механизмы, с помощью которых активированная микроглия и астроциты убивают нейроны, были идентифицированы в клеточной культуре.Эти механизмы включают активацию НАДФН-оксидазы фагоцитов в микроглии и экспрессию iNOS в глии. Эта комбинация вызывает апоптоз посредством продукции ONOO — . Индуцируемый NO, производный от NOS, также действует синергетически с гипоксией, вызывая гибель нейронов, поскольку NO ингибирует цитохромоксидазу. Это может привести к высвобождению глутамата и эксайтотоксичности 57,58 (см. Раздел «Роль нейрональной синтазы оксида азота в патофизиологии, посвященный nNOS и эксайтотоксичности»).
Наконец, чрезмерное производство NO iNOS играет решающую роль в септическом шоке ( Рисунок ).Это состояние характеризуется массивным расширением артериол, гипотонией и повреждением микрососудов. Бактериальные эндотоксины обычно вызывают симптомы. Количество медиаторов, таких как фактор активации тромбоцитов, тромбоксан A 2 , простаноиды и цитокины, такие как интерлейкин-1, фактор некроза опухоли-α и интерферон-γ, повышаются при септическом шоке и участвуют в его патофизиологии. Однако падение артериального давления происходит преимущественно из-за избыточной продукции NO, индуцированной iNOS в стенке сосудов. 59,60
Эндотелиальная синтаза оксида азота
Эндотелиальная БДУ в основном экспрессируется в эндотелиальных клетках ( фигура ). Однако изофермент также был обнаружен в сердечных миоцитах, тромбоцитах, некоторых нейронах головного мозга, в синцитиотрофобластах плаценты человека и в эпителиальных клетках канальцев почки LLC-PK 1 . 28,38
Подобно nNOS, кальмодулин, активированный Ca 2+ , важен для регуляции активности eNOS.Эндотелиальная NOS синтезирует NO пульсирующе, причем активность eNOS заметно увеличивается, когда внутриклеточный Ca 2+ повышается. Ca 2+ индуцирует связывание кальмодулина с ферментом. 20 Однако некоторые другие белки также взаимодействуют с eNOS и регулируют ее активность. Например, было обнаружено, что белок теплового шока 90 (hsp90) связан с eNOS и служит аллостерическим модулятором, активирующим фермент 61 и способствующим (повторному) связыванию eNOS 62,63 (см. Далее в тексте).Фракция eNOS, локализованная в кавеолах 64 , может взаимодействовать с белком оболочки кавеол, кавеолином-1. Кавеолин-1 — тонизирующий ингибитор активности eNOS. Эта концепция была подтверждена генетически, потому что кровеносные сосуды мышей с дефицитом кавеолина-1 демонстрируют усиленную эндотелий-зависимую релаксацию. 65 Механически рекрутирование кальмодулина и hsp90 в eNOS может вытеснять кавеолин-1 из фермента, что приводит к активации фермента. 66
Однако eNOS также может активироваться стимулами, которые не вызывают устойчивого увеличения внутриклеточного Ca 2+ , но все же вызывают длительное высвобождение NO.Лучше всего установленный такой стимул — это напряжение сдвига жидкости. Эта активация опосредуется фосфорилированием фермента. 67,68 Белок eNOS может фосфорилироваться по нескольким остаткам серина (Ser), треонина (Thr) и тирозина (Tyr). Фосфорилирование Ser1177 стимулирует поток электронов внутри домена редуктазы, увеличивает чувствительность фермента к Ca 2+ и представляет собой дополнительный и независимый механизм активации eNOS. 68,69 Эстроген и фактор роста эндотелия сосудов (VEGF) фосфорилируют eNOS в основном через Ser / Thr киназу Akt, инсулин, вероятно, активирует как Akt, так и AMP-активированную протеинкиназу (AMPK), индуцированное брадикинином фосфорилирование Ser1177 опосредуется с помощью Ca 2+ / кальмодулин-зависимой протеинкиназы II (CaMKII), а напряжение сдвига вызывает фосфорилирование, главным образом, за счет активации протеинкиназы A (PKA) ( фигура ).Недавние доказательства использования Akt1-дефицитных мышей, несущих knock-in мутации критического сайта фосфорилирования Akt1 на eNOS, доказали, что киназа Akt1 является критическим регулятором функции eNOS также in vivo . 70 Ser1176 представляет собой сайт фосфорилирования Akt1 у мыши, который соответствует Ser1177 у человека. Фосфомиметическая мутация Ser1176Asp делает фермент конститутивно активным, тогда как мутация Ser1176Ala снижает активность фермента. 70 Таким образом, хотя все упомянутые киназы могут регулировать eNOS Ser1177 in vitro , Akt1 является единственной киназой, которая, как доказано, регулирует функцию eNOS in vivo .
Регулирование активности эндотелиальной NOS посредством внутриклеточного Ca 2+ и фосфорилирования. Увеличение внутриклеточного Ca 2+ приводит к усиленному связыванию кальмодулина (CaM) с ферментом, что, в свою очередь, смещает аутоингибиторную петлю и облегчает поток электронов от НАДФН в домене редуктазы к гему в оксигеназе. домен. Установленными функционально важными сайтами фосфорилирования в эндотелиальной NOS человека являются Ser1177 и Thr495. В покоящихся эндотелиальных клетках Ser1177 обычно не фосфорилируется.Фосфорилирование индуцируется, когда клетки подвергаются воздействию эстрогенов, фактора роста эндотелия сосудов (VEGF), инсулина, брадикинина или напряжения сдвига жидкости. Киназы, ответственные за фосфорилирование (зеленые шестиугольники), зависят от первичного стимула. Эстроген и фактор роста эндотелия сосудов вызывают фосфорилирование Ser1177 путем активации серин / треонинкиназы Akt. Пока что Akt1 является единственной киназой, которая, как было доказано, регулирует функцию эндотелиальной NOS in vivo (обведена зеленым шестиугольником). Инсулин, вероятно, активирует как Akt, так и AMP-активированную протеинкиназу (AMPK), индуцированное брадикинином фосфорилирование Ser1177 опосредуется Ca 2+ / кальмодулин-зависимой протеинкиназой II (CaMKII), а напряжение сдвига приводит к фосфорилированию эндотелия. NOS в основном через протеинкиназу A (PKA).Фосфорилирование остатка Ser1177 увеличивает поток электронов через домен редуктазы и, следовательно, активность фермента. Остаток Thr495 эндотелиальной NOS человека имеет тенденцию постоянно фосфорилироваться в эндотелиальных клетках. Thr495 является негативным регуляторным сайтом, и его фосфорилирование связано со снижением потока электронов и активности фермента. Конститутивно активная киназа, которая фосфорилирует эндотелиальный NOS Thr495, скорее всего, является протеинкиназой C (PKC, желтый шестиугольник). Фосфорилирование Thr495 снижает эндотелиальную активность NOS (желтый блок-стрелка).Фосфатаза, которая дефосфорилирует Thr495, по-видимому, представляет собой протеинфосфатазу1 (PP1, черный флажок с черной стрелкой).
Thr495 имеет тенденцию фосфорилироваться в нестимулированных условиях (скорее всего, протеинкиназой C). Фосфорилирование Thr495, вероятно, мешает связыванию кальмодулина с кальмодулин-связывающим доменом. Фактически, дефосфорилирование Thr495 связано со стимулами, которые повышают внутриклеточные концентрации Ca 2+ и повышают активность eNOS.Когда Thr495 дефосфорилирован, с eNOS связывается значительно больше кальмодулина. 68 Однако было показано, что дефосфорилирование Thr495 способствует разобщению eNOS (см. Ниже). 71
Другие сайты фосфорилирования eNOS человека включают остатки Ser114, Ser633, Tyr81 и Tyr657. Фосфорилирование этих остатков является интенсивно изучаемой областью и может иметь важные последствия для активности ферментов, как было недавно рассмотрено. 72
Физиологические функции эндотелиальной синтазы оксида азота
Расширение сосудов и ингибирование агрегации и адгезии тромбоцитов
Эндотелиальная БДУ является гомеостатическим регулятором многих важных сердечно-сосудистых функций.NO, происходящий из эндотелиальной NOS, расширяет все типы кровеносных сосудов, стимулируя растворимую гуанилилциклазу и увеличивая циклический GMP в гладкомышечных клетках. 3,4,73 Делеция гена eNOS приводит к повышению артериального давления. 74,75 Оксид азота, высвобождаемый в просвет сосудов, является мощным ингибитором агрегации тромбоцитов и адгезии к стенке сосудов. 76–78 Помимо защиты от тромбоза, это также предотвращает высвобождение тромбоцитарных факторов роста, которые стимулируют пролиферацию гладких мышц и производство матричных молекул.Эндотелиальная БДУ также важна для адаптивного ремоделирования сосудов к хроническим изменениям кровотока. 79
Ингибирование адгезии лейкоцитов и сосудистого воспаления
Эндотелиальный NO контролирует экспрессию генов, участвующих в атерогенезе. Оксид азота снижает экспрессию хемоаттрактантного белка MCP-1. 80 Оксид азота может также ингибировать адгезию лейкоцитов к стенке сосуда либо путем нарушения способности молекулы адгезии лейкоцитов CD11 / CD18 связываться с поверхностью эндотелиальных клеток, либо путем подавления экспрессии CD11 / CD18 на лейкоцитах. 81,82 Прилипание лейкоцитов является ранним событием в развитии атеросклероза, и, следовательно, NO может защищать от начала атерогенеза.
Нарушение целостности барьера эндотелиального монослоя может инициировать провоспалительные события. NO, происходящий из эндотелия, предотвращает апоптоз эндотелиальных клеток, вызванный провоспалительными цитокинами и проатеросклеротическими факторами, включая активные формы кислорода (ROS) и ангиотензин II (AT). Подавление апоптоза может также способствовать противовоспалительному и антиатеросклеротическому эффекту NO, полученного из эндотелия. 83
Контроль пролиферации гладких мышц сосудов
Кроме того, было показано, что NO ингибирует синтез ДНК, митогенез и пролиферацию гладкомышечных клеток сосудов. 84–87 Эти антипролиферативные эффекты, вероятно, опосредуются циклическим GMP. 84,85,88 Подавление агрегации и адгезии тромбоцитов защищает гладкие мышцы от воздействия факторов роста, полученных из тромбоцитов. Следовательно, NO также предотвращает более позднюю стадию атерогенеза — образование фиброзных бляшек.Основываясь на комбинации этих эффектов, NO, продуцируемый в эндотелиальных клетках, можно рассматривать как антиатеросклеротический принцип 89 ( фигура ).
Стимуляция ангиогенеза NO, производная эндотелиальной синтазы оксида азота
NO, производная эндотелиальной NOS, играет критическую роль в постнатальном ангиогенезе, опосредуя сигналы ниже ангиогенных факторов. Недавние открытия на eNOS-дефицитных мышах указывают на новую и ранее неизвестную роль NO в развитии легких плода и морфогенезе легких.Легочный фенотип мышей с дефицитом eNOS очень напоминает дисплазию альвеолярных капилляров у людей, форму злокачественной легочной гипертензии у новорожденных, которая проявляется дефектным развитием сосудов легких и респираторным дистрессом. 90 Аналогичным образом было обнаружено, что eNOS имеет решающее значение для образования коллатералей и ангиогенеза постишемии. 91 Кроме того, положительные эффекты NO на выживаемость эндотелиальных клеток, вероятно, также вносят вклад в проангиогенные эффекты NO. 83
Активация эндотелиальных клеток-предшественников оксидом азота, производным от эндотелиальной синтазы оксида азота
У мышей с удаленным геном eNOS наблюдается нарушение неоваскуляризации. Это было связано с дефектом мобилизации клеток-предшественников. Мобилизация эндотелиальных клеток-предшественников с помощью VEGF снижена у мышей с дефицитом eNOS. В модели ишемии задних конечностей у этих мышей внутривенная инфузия клеток-предшественников дикого типа, но не трансплантация костного мозга, может спасти дефектную неоваскуляризацию.Это предполагает, что мобилизация клеток-предшественников из костного мозга нарушена у мышей с дефицитом eNOS. Действительно, матриксная металлопротеиназа-9, которая необходима для мобилизации стволовых клеток, была снижена в костном мозге мышей с дефицитом eNOS. Таким образом, eNOS, экспрессируемая стромальными клетками костного мозга, влияет на рекрутирование стволовых клеток и клеток-предшественников. Таким образом, снижение системной биоактивности NO, наблюдаемое при ишемической болезни сердца, может способствовать нарушению неоваскуляризации. 92
Также у пациентов с ишемической кардиомиопатией мононуклеарные клетки костного мозга демонстрируют пониженную способность к неоваскуляризации in vivo .Как упоминалось выше, NO играет важную роль в неоваскуляризации, и биодоступность NO обычно снижается у пациентов с ишемической болезнью сердца. Предварительная обработка клеток костного мозга этих пациентов усилителем экспрессии и активности eNOS 4-фтор- N -индан-2-илбензамид (AVE9488) 93 значительно увеличила экспрессию и активность eNOS. 94 Это связано с повышенной миграционной способностью клеток костного мозга in vitro и улучшенной способностью к неоваскуляризации этих клеток в модели ишемической задней конечности мыши in vivo .Эта улучшенная перфузия конечностей обработанными AVE9488 клетками костного мозга не опосредована NO, потому что она отменяется предварительной обработкой клеток ингибитором eNOS N G -нитро-1-аргининовым метиловым эфиром. Кроме того, соединение AVE9488 не оказало влияния на нарушенную миграционную способность клеток костного мозга мышей с дефицитом eNOS. Таким образом, фармакологическое усиление экспрессии и активности eNOS, по крайней мере, частично обращает нарушенную функциональную активность клеток костного мозга пациентов с ишемической кардиомиопатией. 94 Аналогичным образом стимулятор eNOS симвастатин (см. Ниже) увеличивал количество функционально активных эндотелиальных клеток-предшественников у пациентов с инфарктом миокарда. 95
Генная терапия синтазой оксида азота
Генная терапия относится к переносу определенного гена в ткань хозяина, чтобы вмешаться в процесс заболевания, что приводит к облегчению симптомов. Генная терапия синтазой оксида азота была в центре внимания многочисленных исследований, поскольку дисфункция этого фермента связана с несколькими типами сердечно-сосудистых заболеваний.Исследования были сосредоточены на эффектах доставки генов изоформ NOS (eNOS, iNOS или nNOS) на животных моделях сосудистого тонуса, ишемии-реперфузионного повреждения, гиперплазии интимы и рестеноза. Во многих доклинических моделях сердечно-сосудистых заболеваний сосудистая доставка генов оказалась терапевтически полезной. Эндотелиальная БДУ представляется особенно многообещающей, поскольку она подавляет гиперплазию интимы и усиливает реэндотелиализацию в поврежденных кровеносных сосудах. Очевидная долгосрочная цель — применить преимущества генной терапии NOS, наблюдаемые на животных моделях, в клиническую практику.Однако на этом пути требуется дальнейшая работа для улучшения систем доставки и сведения к минимуму негативных побочных эффектов. 96,97
Роль эндотелиальной синтазы оксида азота в патофизиологии
Пациенты с сердечно-сосудистыми факторами риска (такими как гипертония, гиперхолестеринемия, сахарный диабет, курение сигарет и т. Д.) И пациенты с сосудистыми заболеваниями демонстрируют эндотелиальную дисфункцию, т.е. эндотелия для выработки адекватного количества биоактивного NO (и для производства NO-опосредованной вазодилатации).Факторы риска сердечно-сосудистых заболеваний и сосудистые заболевания также связаны с повышенным производством АФК. Существует несколько ферментных систем, которые потенциально могут продуцировать АФК в стенке сосуда. К ним относятся НАДФН-оксидазы, ксантиноксидаза, ферменты дыхательной цепи митохондрий и несвязанные eNOS (см. Ниже). 98 Из них НАДФН-оксидазы имеют первостепенное значение для образования АФК ( Рисунок A ). В стенке сосудов существует несколько изоформ НАДФН-оксидазы, продуцирующей O 2 — • .Они экспрессируются в эндотелиальных и гладкомышечных клетках, а также в адвентиции.
Возможные механизмы, с помощью которых факторы риска сердечно-сосудистых заболеваний приводят к окислительному стрессу и разъединению эндотелиальной БДУ. ( A ) При многих типах сосудистых заболеваний НАДФН-оксидаза активируется в сосудистой стенке и генерирует супероксид (O 2 — • ). Было показано, что при экспериментальном сахарном диабете и гипертензии, вызванной ангиотензином II, это опосредуется протеинкиназой C (PKC). 168,169 Экспрессия эндотелиальной NOS также увеличивается при сосудистых заболеваниях. H 2 O 2 , продукт дисмутации O 2 — • может увеличивать экспрессию эндотелиальной NOS посредством транскрипционных и посттранскрипционных механизмов (SOD, супероксиддисмутаза). 170 Кроме того, активация протеинкиназы C может усиливать экспрессию эндотелиальной NOS, 171 и ингибиторы протеинкиназы C снижают уровни экспрессии эндотелиальной NOS при сосудистых заболеваниях. 169 Продукты НАДФН-оксидаз и эндотелиальных NOS, O 2 — • и NO · быстро рекомбинируют с образованием пероксинитрита (ONOO — ). Это может окислять важный кофактор эндотелиальной NOS (6 R -) 5,6,7,8-тетрагидробиоптерин (BH 4 ) до радикала тригидробиоптерина (BH 3 • ). 172,173 BH 3 • может непропорционально хиноноиду 6,7- [8H] -H 2 -биоптерину (BH 2 ).Как следствие, восстановление кислорода и O 2 эндотелиальной NOS не связано с образованием NO · , и функциональная NOS превращается в дисфункциональный O 2 — • -производящий фермент, который способствует окислительному стрессу сосудов. . Повышенная экспрессия эндотелиальной NOS (см. Выше) усугубляет ситуацию. ( B ) Окисление BH 4 до биологически неактивных продуктов, таких как радикал BH 3 • или BH 2 , также снижает сродство субстрата l-аргинина (l-Arg) к NO и NOS катализирует несвязанное восстановление O 2 , что приводит к образованию O 2 — • (и, возможно, также H 2 O 2 ).
Молекулярная основа эндотелиальной дисфункции при сосудистых заболеваниях: инактивация биоактивного оксида азота и разобщение эндотелиальной синтазы оксида азота
Из-за повышенного окислительного стресса, наблюдаемого при сосудистых заболеваниях, повышенная деградация NO в результате его реакции с O 2 — • произойдет. Однако также было показано, что окислительный стресс превращает eNOS из фермента, продуцирующего NO, в фермент, который вырабатывает O 2 — • . Этот процесс был назван NOS-разъединением (, рисунок B, ).Механизмы, участвующие в разобщении eNOS, включают окисление критического кофактора NOS BH 4 , истощение L-аргинина и накопление эндогенных метиларгининов. Совсем недавно S-глутатионилирование eNOS было предложено в качестве еще одного механизма, ведущего к разобщению eNOS.
Гипотеза (6
R -) 5,6,7,8-тетрагидробиоптеринаКак подробно описано выше, функциональная eNOS переносит электроны от НАДФН через флавины FAD и FMN в гем, где субстрат l-аргинин окисляется до l-цитруллина и NO. 99 Продукт реакции NO и O 2 — • , ONOO — , может отделить восстановление кислорода от образования NO в NOS. Окисление или удаление необходимого кофактора BH 4 99-101 или окислительное повреждение цинк-тиолатного кластера, участвующего в BH 4 и связывание l-аргинина 102 , может быть причиной разобщения eNOS в этой ситуации ( Рисунок A и B ). ONOO — может окислять BH 4 до биологически неактивного радикала BH 3 • , который может диспропорционировать хиноноиду 6,7- [8H] -H 2 -биоптерину. 103,104 Было показано, что продукция NO посредством eNOS тесно коррелирует с внутриклеточной концентрацией BH 4 , 105,106 и BH 4 Уровни были снижены во многих моделях сердечно-сосудистых заболеваний 107–109,101 и в пациенты с эндотелиальной дисфункцией. 110–112
Обеспечение l-аргинином эндотелиальной синтазы оксида азота и разобщение эндотелиальной синтазы оксида азота
В патофизиологии животных и человека (гиперхолестеринемия и гипертензия) добавление l-аргинина может улучшить эндотелиальную дисфункцию. 113–116 Нормальные концентрации l-аргинина в плазме составляют ∼100 мкмоль / л. Даже в патофизиологии они почти не опускаются ниже 60 мкмоль / л, а в клетках наблюдается до 10-кратное накопление l-аргинина. 117 С другой стороны, K m eNOS для l-аргинина составляет всего ∼3 мкмоль / л. 118 Кроме того, эндотелиальные клетки человека даже не зависят от поглощения l-аргинина из внеклеточного пространства; они могут эффективно рециркулировать l-цитруллин в l-аргинин, а также могут получать l-аргинин в результате протеолиза. 119,120
Однако эндотелиальные клетки экспрессируют аргиназы, которые могут конкурировать с eNOS за субстрат и, в случае высокой экспрессии, «голодать» eNOS. 121–124 Относительный дефицит l-аргинина в непосредственной близости от eNOS, вызванный чрезмерной активностью аргиназы, возможен и может частично объяснять положительные эффекты добавок l-аргинина. Этим эффектам также могут способствовать не субстратные эффекты l-аргинина. К ним относятся потенциальные свойства прямого улавливания радикалов гуанидиновой азотной группы, кооперативность между l-аргинином и сайтами связывания BH 4 NOS, 125,126 или конкуренция l-аргинина с асимметричным диметил-l-аргинином (ADMA ). 127
Разъединение асимметричного диметил-1-аргинина и эндотелиальной синтазы оксида азота
Асимметричный диметил-1-аргинин считается фактором риска общей сердечно-сосудистой смертности. 128 Асимметричный диметил-1-аргинин является эндогенным ингибитором eNOS, но повышенный уровень ADMA также связан с разобщением eNOS. 129 Активность (не экспрессия) ключевого фермента для продукции ADMA, протеина аргинин N -метилтрансферазы (PRMT, тип I), 130 и фермента, разрушающего ADMA, диметиларгининдиметиламиногидролазы (DDAH) 131 чувствительны к окислительно-восстановительному потенциалу.В различных моделях было показано, что окислительный стресс увеличивает активность PRMT (s) и снижает активность DDAH, что приводит к увеличению концентрации ADMA. 127 130 131 Таким образом, увеличение производства ROS может вызвать повышение уровней ADMA.
S-глутатионилирование эндотелиальной синтазы оксида азота: еще один механизм, приводящий к разъединению эндотелиальной синтазы оксида азота
При нескольких болезненных состояниях, связанных с окислительным стрессом (см. Выше), добавка BH 4 только частично восстанавливает функциональность eNOS.Остатки цистеина важны для функции eNOS. Белковые тиолы могут подвергаться S-глутатионилированию, модификации белка, участвующей в передаче сигналов в клетке. Условия окислительного стресса способствуют S-глутатионилированию белков. S-глутатионилирование eNOS обратимо снижает продукцию NO и увеличивает образование O 2 — • , главным образом, из домена редуктазы. Два высококонсервативных остатка цистеина в домене редуктазы были идентифицированы как сайты S-глутатионилирования. 132 Эндотелиальная БДУ S-глутатионилирование в эндотелиальных клетках сопровождается нарушением эндотелий-зависимой вазодилатации. В кровеносных сосудах животных с гипертонией повышено S-глутатионилирование eNOS и снижено опосредованное эндотелием расширение сосудов. Это состояние отменяется тиол-специфическими восстанавливающими агентами, которые обращают S-глутатионилирование. Таким образом, S-глутатионилирование eNOS, вероятно, представляет собой дополнительный механизм, участвующий в разобщении eNOS. 132,133
Плейотропное действие обычных сердечно-сосудистых препаратов, улучшающих функцию эндотелия
Препараты, влияющие на систему ренин-ангиотензин-альдостерон, и статины (ингибиторы 3-гидрокси-3-метилгултарил-коэнзима А-редуктазы) способны предотвращать дисфункцию эндотелия и дисфункцию эндотелия. Отключение eNOS.
Лекарства, влияющие на систему ренин-ангиотензин-альдостерон
Некоторые компоненты системы ренин-ангиотензин-альдостерон активируются в атеросклеротических сосудах. И ангиотензин II, и альдостерон способствуют эндотелиальной дисфункции. 134 Ангиотензин II активирует НАДФН-оксидазу посредством стимуляции AT 1 . 135 Кроме того, рецептор AT 1 активируется in vitro липопротеином низкой плотности. 136
У кроликов с наследственной гиперлипидемией Ватанабе ингибитор ренина алискирен увеличивает экспрессию eNOS, усиливает фосфорилирование eNOS по Ser1177 (тем самым увеличивая активность), снижает экспрессию NADPH-оксидазы, увеличивает сосудистые уровни BH 4 и восстанавливает разобщение eNOS. 137
Ингибиторы ангиотензин-превращающего фермента и блокаторы рецепторов AT 1 обладают непрямым антиоксидантным действием, предотвращая активацию НАДФН-оксидазы. 138–141 Кроме того, они также могут увеличивать активность внеклеточной супероксиддисмутазы (SOD3). 142 Ингибиторы ангиотензин-превращающего фермента значительно снижают сердечно-сосудистые события у пациентов с установленной ишемической болезнью сердца или с высоким риском этого заболевания. 143 Блокатор рецепторов AT 1 лозартан восстанавливает выработку NO в клубочках за счет увеличения экспрессии белка GTP циклогидролазы1 (ограничивающего скорость фермента для синтеза BH 4 ) и повышения уровней BH 4 у диабетических крыс. 144
Селективный антагонист альдостерона эплеренон и эналаприл снижают активность НАДФН-оксидазы, повышают уровень BH в сосудах 4 и повышают экспрессию eNOS и биодоступность NO. Эплеренон также увеличивает фосфорилирование eNOS по Ser1177. Оба препарата уменьшают образование атеросклеротических бляшек. 134
Эти плейотропные эффекты соединений, влияющие на систему ренин-ангиотензин-альдостерон, могут вносить значительный вклад в терапевтический эффект таких лекарств.
Статины
Снижающие холестерин статины обладают дополнительным холестерин-независимым или плейотропным действием при сердечно-сосудистых заболеваниях. 145 Эти свойства включают улучшение функции эндотелия, стабилизацию атеросклеротических бляшек, ингибирование окислительного стресса и воспаления и снижение тромбогенных реакций. 146 Эти эффекты статинов частично опосредованы действием на eNOS, поскольку они могут подавляться ингибиторами eNOS 147 и отсутствуют у мышей с дефицитом eNOS. 95
Статины увеличивают экспрессию eNOS, 148 , но также повышают активность eNOS за счет уменьшения содержания кавеолина 149 и активации пути фосфатидилинозитол-3-киназа / Akt. 150
Некоторые статины ингибируют эндотелиальное образование O 2 — • за счет снижения экспрессии и / или активности НАДФН-оксидазы и предотвращения изопренилирования p21 Rac, что имеет решающее значение для функциональной НАДФН-оксидазы. 151 Кроме того, симвастатин более чем удваивает активность SOD3.
Статины также увеличивают экспрессию мРНК GTP циклогидролазы1 в эндотелиальных клетках и повышают внутриклеточные уровни BH 4 . 152 Было показано, что аторвастатин нормализует функцию эндотелия и снижает окислительный стресс за счет ингибирования сосудистых НАДФН-оксидаз и предотвращения разобщения eNOS за счет активации GTP-циклогидролазы1. 153 Вместе эти эффекты могут способствовать антиатерогенному действию статинов. 154,155
Выводы
Все три изофермента NOS выполняют регуляторные функции в сердечно-сосудистой системе. Нейрональные NOS участвуют в центральной регуляции артериального давления, а nNOS-содержащие (нитрергические) нервы могут расширять определенные сосудистые русла. Нитрергические нервы имеют особое значение для расслабления кавернозного тела и эрекции полового члена. Для действия ингибиторов фосфодиэстеразы 5 требуется по крайней мере остаточная активность nNOS. Индуцируемая NOS экспрессируется в атеросклеротической бляшке и является важным медиатором падения артериального давления при септическом шоке.Наиболее важной изоформой является eNOS, которая поддерживает расширение кровеносных сосудов, контролирует кровяное давление и обладает множеством других вазопротекторных и антиатеросклеротических эффектов. Хотя нет никаких доказательств того, что eNOS является «геном болезни», многие факторы риска сердечно-сосудистых заболеваний приводят к окислительному стрессу, разобщению eNOS и эндотелиальной дисфункции в сосудистой сети. Лекарства, влияющие на систему ренин-ангиотензин-альдостерон, а также статины полезны для предотвращения эндотелиальной дисфункции. Дальнейшее выяснение того, как эти терапевтические агенты способствуют связыванию eNOS в условиях повышенного окислительного стресса, может дать представление о других потенциальных путях, ведущих к благоприятному действию NO в сердечно-сосудистой системе.
Финансирование
Оригинальная работа наших лабораторий, участвующих в этом обзоре, была поддержана Комплексным исследовательским и лечебным центром «Тромбоз и гемостаз» Федерального министерства образования и исследований Германии (BMBF) грантом LI-1042 / 1-1. от Немецкого исследовательского фонда (Deutsche Forschungsgemeinschaft) и грантами R01 HL64793;, R01 HL61371;, R01 HL081190;, R01 HL096670; и P01 HL70295 Национальных институтов здравоохранения США.
Конфликт интересов: Не объявлен.
Ссылки
1. О’Делл Т.Дж., Хокинс Р.Д., Кандел Э.Р., Арансио О. Тесты роли двух диффундирующих веществ в долговременной потенциации: данные о оксиде азота как возможном раннем ретроградном посланнике. Proc Natl Acad Sci USA. 1991; 88: 11285–11289. [Бесплатная статья PMC] [PubMed] [Google Scholar] 2. Шуман Э.М., Мэдисон Д.В. Необходимость в оксиде азота межклеточного посредника при долговременной потенциации. Наука. 1991; 254: 1503–1506. [PubMed] [Google Scholar] 3. Рапопорт Р.М., Дразнин М.Б., Мурад Ф.Эндотелий-зависимая релаксация в аорте крысы может опосредоваться циклическим GMP-зависимым фосфорилированием белков. Природа. 1983; 306: 174–176. [PubMed] [Google Scholar] 4. Förstermann U, Mülsch A, Böhme E, Busse R. Стимуляция растворимой гуанилатциклазы ацетилхолин-индуцированным фактором, полученным из эндотелия из артерий кроликов и собак. Circ Res. 1986; 58: 531–538. [PubMed] [Google Scholar] 5. Хан Б.В., Харрисон Д.Г., Ольбрых М.Т., Александр Р.В., Медфорд Р.М. Оксид азота регулирует экспрессию гена молекулы 1 адгезии сосудов и окислительно-восстановительные процессы транскрипции в эндотелиальных клетках сосудов человека.Proc Natl Acad Sci USA. 1996; 93: 9114–9119. [Бесплатная статья PMC] [PubMed] [Google Scholar] 6. Гуди Т., Хонг Г.К., Ваандрагер А.Б., Ломанн С.М., Пильц РБ. Оксид азота и цГМФ регулируют экспрессию генов в нейрональных и глиальных клетках путем активации цГМФ-зависимой протеинкиназы II типа. FASEB J. 1999; 13: 2143–2152. [PubMed] [Google Scholar] 7. Пантопулос К., Хентце М.В. Передача сигналов оксида азота железо-регуляторному белку: прямой контроль трансляции мРНК ферритина и стабильности мРНК рецептора трансферрина в трансфицированных фибробластах.Proc Natl Acad Sci USA. 1995; 92: 1267–1271. [Бесплатная статья PMC] [PubMed] [Google Scholar] 8. Лю XB, Hill P, Haile DJ. Роль ферропортина, чувствительного к железу элемента в регуляции генов, зависимых от железа и оксида азота. Blood Cells Mol Dis. 2002. 29: 315–326. [PubMed] [Google Scholar] 9. Поздняков Н., Ллойд А., Редди В. Н., Ситарамайя А. Эндогенное АДФ-рибозилирование белков наружного сегмента палочек, регулируемое оксидом азота. Biochem Biophys Res Commun. 1993; 192: 610–615. [PubMed] [Google Scholar] 10. Брюн Б., Диммелер С, Молина и Ведиа Л., Лапетина Э.Г.Оксид азота: сигнал для ADP-рибозилирования белков. Life Sci. 1994; 54: 61–70. [PubMed] [Google Scholar] 11. Миккельсен Р.Б., Вардман П. Биологическая химия реактивного кислорода и азота и механизмы радиационной передачи сигналов. Онкоген. 2003; 22: 5734–5754. [PubMed] [Google Scholar] 12. Ли Дж. Х., Ян Э. С., Пак Дж. У. Инактивация НАДФ + -зависимой изоцитратдегидрогеназы пероксинитритом. Последствия цитотоксичности и вызванного алкоголем поражения печени. J Biol Chem. 2003. 278: 51360–51371.[PubMed] [Google Scholar] 13. Риднур Л.А., Томас Д.Д., Манкарди Д., Эспи М.Г., Миранда К.М., Паолоччи Н., Филиш М., Фукуто Дж., Винк Д.А. Химия нитрозативного стресса, вызванного оксидом азота и реактивными формами оксида азота. Взгляд на стрессовые биологические ситуации. Biol Chem. 2004; 385: 1–10. [PubMed] [Google Scholar] 14. Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Структура димера оксигеназы синтазы оксида азота с птерином и субстратом. Наука. 1998. 279: 2121–2126.[PubMed] [Google Scholar] 16. Нишимура Дж. С., Мартасек П., Макмиллан К., Салерно Дж., Лю К., Гросс СС, Мастерс Б.С. Модульная структура нейрональной синтазы оксида азота: локализация сайта связывания аргинина и модуляция птерином. Biochem Biophys Res Commun. 1995; 210: 288–294. [PubMed] [Google Scholar] 17. Ноубл М.А., Манро А.В., Риверс С.Л., Робледо Л., Дафф С.Н., Йеллоулис Л.Дж., Симидзу Т., Сагами И., Гиллеметт Д.Г., Чепмен С.К. Потенциометрический анализ флавиновых кофакторов нейрональной синтазы оксида азота.Биохимия. 1999; 38: 16413–16418. [PubMed] [Google Scholar] 18. Stuehr D, Pou S, Rosen GM. Восстановление кислорода синтазами оксида азота. J Biol Chem. 2001; 276: 14533–14536. [PubMed] [Google Scholar] 19. Cho HJ, Xie QW, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Nathan C. Кальмодулин является субъединицей синтазы оксида азота из макрофагов. J Exp Med. 1992; 176: 599–604. [Бесплатная статья PMC] [PubMed] [Google Scholar] 20. Хемменс Б., Майер Б. Энзимология синтаз оксида азота. Методы Мол биол. 1998; 100: 1–32.[PubMed] [Google Scholar] 21. Хемменс Б., Гесслер В., Шмидт К., Майер Б. Роль связанного цинка в стабилизации димера, но не ферментативная активность нейрональной синтазы оксида азота. J Biol Chem. 2000; 275: 35786–35791. [PubMed] [Google Scholar] 22. Раман К.С., Ли Х., Марташек П., Краль В., Мастерс Б.С., Поулос Т.Л. Кристаллическая структура конститутивной эндотелиальной синтазы оксида азота: парадигма функции птерина с участием нового металлического центра. Клетка. 1998. 95: 939–950. [PubMed] [Google Scholar] 23. Li H, Raman CS, Glaser CB, Blasko E, Young TA, Parkinson JF, Whitlow M, Poulos TL.Кристаллические структуры не содержащего цинка и связанного гемом домена индуцибельной синтазы оксида азота человека. Значение для стабильности димера и сравнение с эндотелиальной синтазой оксида азота. J Biol Chem. 1999; 274: 21276–21284. [PubMed] [Google Scholar] 24. Furchgott RF, Cherry PD, Zawadzki JV, Jothianandan D. Эндотелиальные клетки как медиаторы вазодилатации артерий. J Cardiovasc Pharmacol. 1984. 53: 557–573. [PubMed] [Google Scholar] 25. Ноулз Р.Г., Паласиос М., Палмер Р.М., Монкада С. Образование оксида азота из l-аргинина в центральной нервной системе: механизм трансдукции для стимуляции растворимой гуанилатциклазы.Proc Natl Acad Sci USA. 1989; 86: 5159–5162. [Бесплатная статья PMC] [PubMed] [Google Scholar] 26. Garthwaite J. Глутамат, оксид азота и межклеточная передача сигналов в нервной системе. Trends Neurosci. 1991; 14: 60–67. [PubMed] [Google Scholar] 27. Zhou L, Zhu DY. Нейрональная синтаза оксида азота: структура, субклеточная локализация, регуляция и клиническое значение. Оксид азота. 2009. 20: 223–230. [PubMed] [Google Scholar] 28. Förstermann U, Closs EI, Pollock JS, Nakane M, Schwarz P, Gath I, Kleinert H. Изоферменты синтазы оксида азота.Характеристика, очистка, молекулярное клонирование и функции. Гипертония. 1994; 23: 1121–1131. [PubMed] [Google Scholar] 29. Nakane M, Schmidt HH, Pollock JS, Förstermann U, Murad F. Клонированная синтаза оксида азота человеческого мозга высоко экспрессируется в скелетных мышцах. FEBS Lett. 1993; 316: 175–180. [PubMed] [Google Scholar] 30. Идзуми Ю., Клиффорд Д. Б., Зорумски К. Ф. Ингибирование долгосрочного потенцирования за счет высвобождения оксида азота, опосредованного NMDA. Наука. 1992; 257: 1273–1276. [PubMed] [Google Scholar] 31. Изуми Ю., Зорумски К.Ф.Оксид азота и длительная синаптическая депрессия в гиппокампе крыс. Нейроотчет. 1993; 4: 1131–1134. [PubMed] [Google Scholar] 32. Holscher C, Rose SP. Ингибитор синтеза оксида азота предотвращает формирование памяти у цыпленка. Neurosci Lett. 1992. 145: 165–167. [PubMed] [Google Scholar] 33. Боме Г.А., Бон С., Лемер М., Рейбо М., Пиот О., Штутцманн Дж. М., Добл А., Бланшар Дж. Измененная синаптическая пластичность и формирование памяти у крыс, получавших ингибитор синтазы оксида азота. Proc Natl Acad Sci USA. 1993; 90: 9191–9194.[Бесплатная статья PMC] [PubMed] [Google Scholar] 34. Togashi H, Sakuma I, Yoshioka M, Kobayashi T, Yasuda H, Kitabatake A, Saito H, Gross SS, Levi R. Действие оксида азота на центральную нервную систему в регуляции артериального давления. J Pharmacol Exp Ther. 1992; 262: 343–347. [PubMed] [Google Scholar] 35. Сакума I, Тогаши Х., Йошиока М., Сайто Х., Янагида М., Тамура М., Кобаяши Т., Ясуда Х., Гросс С.С., Леви Р. NG-метил-1-аргинин, ингибитор синтеза оксида азота, производного из L-аргинина, стимулирует активность симпатического нерва почек in vivo .Роль оксида азота в центральной регуляции симпатического тонуса? Circ Res. 1992; 70: 607–611. [PubMed] [Google Scholar] 36. Элькариб А.О., Шенг Дж.Дж., Бец А.Л., Малвин Р.Л. Центральные эффекты ингибитора синтазы оксида азота ( N -омега-нитро-1-аргинин) на артериальное давление и ренин плазмы. Clin Exp Гипертония. 1993; 15: 819–832. [PubMed] [Google Scholar] 37. Тода Н, Аяджики К., Окамура Т. Контроль системного и легочного артериального давления с помощью оксида азота, образующегося через нейрональную синтазу оксида азота.J Hypertens. 2009; 27: 1929–1940. [PubMed] [Google Scholar] 38. Förstermann U. Регулирование экспрессии и активности синтазы оксида азота. В: Майер Б., редактор. Справочник по экспериментальной фармакологии — оксид азота. Берлин: Спрингер; 2000. С. 71–91. [Google Scholar] 39. Меликян Н., Седдон, доктор медицины, Касадей Б., Човенчик П.Дж., Шах А.М. Нейрональная синтаза оксида азота и регуляция сосудов человека. Trends Cardiovasc Med. 2009. 19: 256–262. [Бесплатная статья PMC] [PubMed] [Google Scholar] 40. Schwarz PM, Kleinert H, Förstermann U.Потенциальное функциональное значение синтазы оксида азота I мозгового и мышечного типа, экспрессируемой в адвентиции и средах аорты крысы. Артериосклер Thromb Vasc Biol. 1999; 19: 2584–2590. [PubMed] [Google Scholar] 41. Kim N, Azadzoi KM, Goldstein I., Saenz de Tejada I. Фактор, подобный оксиду азота, опосредует неадренергическое-нехолинергическое нейрогенное расслабление гладких мышц кавернозного тела полового члена. J Clin Invest. 1991; 88: 112–118. [Бесплатная статья PMC] [PubMed] [Google Scholar] 42. Райфер Дж., Аронсон В. Дж., Буш П. А., Дори Ф. Дж., Игнарро Л. Дж..Оксид азота как медиатор расслабления кавернозного тела в ответ на неадренергическую, нехолинергическую нейротрансмиссию. N Engl J Med. 1992; 326: 90–94. [PubMed] [Google Scholar] 43. Турко И.В., Баллард С.А., Фрэнсис С.Х., Корбин Дж.Д. Ингибирование циклической GMP-связывающей циклической GMP-специфической фосфодиэстеразы (Тип 5) силденафилом и родственными соединениями. Mol Pharmacol. 1999; 56: 124–130. [PubMed] [Google Scholar] 44. Розен RC, Костис JB. Обзор ингибирования фосфодиэстеразы 5 при эректильной дисфункции.Am J Cardiol. 2003; 92: 9М – 18М. [PubMed] [Google Scholar] 45. Штайнерт Дж. Р., Чернова Т., Форсайт ИД. Передача сигналов оксида азота в функции мозга, дисфункции и деменции. Невролог. 2010. 16: 435–452. [PubMed] [Google Scholar] 46. Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. Основанный на окислительно-восстановительном потенциале механизм нейропротекторного и нейродеструктивного действия оксида азота и родственных нитрозосоединений. Природа. 1993; 364: 626–632. [PubMed] [Google Scholar] 47. Браун GC.Оксид азота и гибель нейронов. Оксид азота. 2010. 23: 153–165. [PubMed] [Google Scholar] 48. Тёттруп А., Свейн Д., Форман А. Оксид азота, опосредующий ингибирование NANC в нижнем пищеводном сфинктере опоссума. Am J Physiol. 1991; 260: G385 – G389. [PubMed] [Google Scholar] 49. Лефевр РА. Фармакологическая характеристика нитрергической иннервации желудка. Verh K Acad Geneeskd Belg. 2002. 64: 151–166. [PubMed] [Google Scholar] 50. Натан К.Ф., Хиббс Дж.Б. Роль синтеза оксида азота в антимикробной активности макрофагов.Curr Opin Immunol. 1991; 3: 65–70. [PubMed] [Google Scholar] 51. Винк Д.А., Каспрзак К.С., Марагос К.М., Элеспуру Р.К., Мисра М., Дунамс ТМ, Цебула Т.А., Кох У.Х., Эндрюс А.В., Аллен Дж.С., Кифер Дж.К. Дезаминирующая способность ДНК и генотоксичность оксида азота и его предшественников. Наука. 1991; 254: 1001–1003. [PubMed] [Google Scholar] 52. Фехсел К., Ялови А., Ци С., Буркарт В., Хартманн Б., Колб Х. ДНК островковых клеток является мишенью воспалительной атаки оксида азота. Диабет. 1993; 42: 496–500. [PubMed] [Google Scholar] 53. Ли Л.М., Килборн Р.Г., Адамс Дж., Фидлер И.Дж.Роль оксида азота в лизисе опухолевых клеток цитокин-активированными эндотелиальными клетками. Cancer Res. 1991; 51: 2531–2535. [PubMed] [Google Scholar] 54. Грин SJ, Mellouk S, Hoffman SL, Meltzer MS, Nacy CA. Клеточные механизмы неспецифического иммунитета к внутриклеточной инфекции: цитокин-индуцированный синтез токсичных оксидов азота из l-аргинина макрофагами и гепатоцитами. Immunol Lett. 1990; 25: 15–19. [PubMed] [Google Scholar] 55. Kröncke KD, Kolb-Bachofen V, Berschick B, Burkart V, Kolb H. Активированные макрофаги убивают сингенные островковые клетки поджелудочной железы посредством аргинин-зависимого образования оксида азота.Biochem Biophys Res Commun. 1991; 175: 752–758. [PubMed] [Google Scholar] 56. Langrehr JM, Hoffman RA, Billiar TR, Lee KK, Schraut WH, Simmons RL. Синтез оксида азота в ответе аллотрансплантата in vivo : возможный регуляторный механизм. Операция. 1991; 110: 335–342. [PubMed] [Google Scholar] 57. Kanwar JR, Kanwar RK, Burrow H, Baratchi S. Последние достижения в области роли NO при раке и хронических воспалительных заболеваниях. Curr Med Chem. 2009; 16: 2373–2394. [PubMed] [Google Scholar] 58. Браун GC, Neher JJ.Воспалительная нейродегенерация и механизмы микроглиальной гибели нейронов. Mol Neurobiol. 2010. 41: 242–247. [PubMed] [Google Scholar] 59. Макмикинг Дж. Д., Натан С., Хом Дж., Чартрейн Н., Флетчер Д. С., Трумбауэр М., Стивенс К., Се К. В., Сокол К., Хатчинсон Н. и др. Измененные реакции на бактериальную инфекцию и эндотоксический шок у мышей, лишенных индуцибельной синтазы оксида азота. Клетка. 1995. 81: 641–650. [PubMed] [Google Scholar] 60. Ланге М., Энхбаатар П., Накано Ю., Трабер Д.Л. Роль оксида азота в шоке: взгляд на крупные животные.Передние биоски. 2009; 14: 1979–1989. [PubMed] [Google Scholar] 61. Гарсия-Кардена Г., Фан Р., Шах В., Соррентино Р., Чирино Г., Папапетропулос А., Сесса В. Динамическая активация эндотелиальной синтазы оксида азота Hsp90. Природа. 1998; 392: 821–824. [PubMed] [Google Scholar] 62. Причард К.А., младший, Акерман А.В., Гросс ER, Степп Д.В., Ши Й., Фонтана Д.Т., Бейкер Дж.Э., Сесса В. Белок теплового шока 90 обеспечивает баланс оксида азота и супероксид-аниона эндотелиальной синтазы оксида азота. J Biol Chem. 2001; 276: 17621–17624.[PubMed] [Google Scholar] 63. Song Y, Cardounel AJ, Zweier JL, Xia Y. Ингибирование образования супероксида из нейрональной синтазы оксида азота белком теплового шока 90: последствия для регуляции NOS. Биохимия. 2002; 41: 10616–10622. [PubMed] [Google Scholar] 64. Sowa G, Pypaert M, Sessa WC. Различие между сигнальными механизмами в липидных рафтах и кавеолах. Proc Natl Acad Sci USA. 2001; 98: 14072–14077. [Бесплатная статья PMC] [PubMed] [Google Scholar] 65. Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV.Утрата кавеол, сосудистая дисфункция и легочные дефекты у мышей с нарушенным геном кавеолина-1. Наука. 2001; 293: 2449–2452. [PubMed] [Google Scholar] 66. Граттон Дж. П., Фонтана Дж., О’Коннор Д. С., Гарсия-Кардена Дж., МакКейб Т. Дж., Сесса В. Восстановление эндотелиальной синтазы оксида азота (eNOS), hsp90 и комплекса кавеолина-1 in vitro . Доказательства того, что hsp90 способствует вытеснению eNOS из кавеолина-1, стимулированного кальмодулином. J Biol Chem. 2000; 275: 22268–22272. [PubMed] [Google Scholar] 67.Фултон Д., Грэттон Дж. П., Маккейб Т. Дж., Фонтана Дж., Фуджио Ю., Уолш К., Франке Т.Ф., Папапетропулос А., Сесса В. Регулирование производства оксида азота эндотелием протеинкиназой Akt. Природа. 1999; 399: 597–601. [Бесплатная статья PMC] [PubMed] [Google Scholar] 68. Флеминг И., Буссе Р. Молекулярные механизмы, участвующие в регуляции эндотелиальной синтазы оксида азота. Am J Physiol Regul Integr Comp Physiol. 2003; 284: R1 – R12. [PubMed] [Google Scholar] 69. Маккейб Т.Дж., Фултон Д., Роман Л.Дж., Сесса В.К. Усиленный поток электронов и сниженная диссоциация кальмодулина могут объяснить «независимую от кальция» активацию eNOS фосфорилированием.J Biol Chem. 2000. 275: 6123–6128. [PubMed] [Google Scholar] 70. Шлейхер М., Ю. Дж., Мурата Т., Дерахшан Б., Аточин Д., Цянь Л., Кашиваги С., Ди Лоренцо А., Харрисон К. Д., Хуанг П. Л., Сесса В. Ось Akt1-eNOS иллюстрирует специфичность отношений киназа-субстрат in vivo . Sci Signal. 2009; 2: ра41. [Бесплатная статья PMC] [PubMed] [Google Scholar] 71. Лин М.И., Фултон Д., Бэббит Р., Флеминг И., Буссе Р., Причард К.А., младший, Сесса В. Фосфорилирование треонина 497 в эндотелиальной синтазе оксида азота координирует взаимодействие метаболизма l-аргинина с эффективным производством оксида азота.J Biol Chem. 2003; 278: 44719–44726. [PubMed] [Google Scholar] 72. Флеминг И. Молекулярные механизмы, лежащие в основе активации eNOS. Pflugers Arch. 2010; 459: 793–806. [PubMed] [Google Scholar] 73. Игнарро Л.Дж., Харбисон Р.Г., Вуд К.С., Кадовиц П.Дж. Активация очищенной растворимой гуанилатциклазы расслабляющим фактором эндотелия из внутрилегочной артерии и вены: стимуляция ацетилхолином, брадикинином и арахидоновой кислотой. J Pharmacol Exp Ther. 1986; 237: 893–900. [PubMed] [Google Scholar] 74. Шесели Э. Г., Маэда Н., Ким Х. С., Десаи К. М., Креге Дж. Х., Лаубах В. Э., Шерман П. А., Сесса В. К., Смитис О.Повышенное артериальное давление у мышей, лишенных эндотелиальной синтазы оксида азота. Proc Natl Acad Sci USA. 1996; 93: 13176–13181. [Бесплатная статья PMC] [PubMed] [Google Scholar] 75. Хуанг П.Л., Хуанг З., Машимо Х., Блох К.Д., Московиц М.А., Беван Дж. А., Фишман М.С.. Гипертония у мышей, лишенных гена эндотелиальной синтазы оксида азота. Природа. 1995; 377: 239–242. [PubMed] [Google Scholar] 76. Alheid U, Frölich JC, Förstermann U. Расслабляющий фактор, полученный из эндотелия из культивированных эндотелиальных клеток человека, ингибирует агрегацию тромбоцитов человека.Thromb Res. 1987. 47: 561–571. [PubMed] [Google Scholar] 77. Busse R, Luckhoff A, Bassenge E. Эндотелиевый релаксантный фактор подавляет активацию тромбоцитов. Naunyn Schmiedebergs Arch Pharmacol. 1987. 336: 566–571. [PubMed] [Google Scholar] 78. Радомский М.В., Палмер Р.М., Монкада С. Антиагрегантные свойства сосудистого эндотелия: взаимодействия между простациклином и оксидом азота. Br J Pharmacol. 1987. 92: 639–646. [Бесплатная статья PMC] [PubMed] [Google Scholar] 79. Рудик Р.Д., Шесели Э.Г., Маеда Н., Смитис О., Сегал С.С., Сесса В.Прямые доказательства важности оксида азота эндотелия в ремоделировании сосудов. J Clin Invest. 1998. 101: 731–736. [Бесплатная статья PMC] [PubMed] [Google Scholar] 80. Zeiher AM, Fisslthaler B, Schray-Utz B, Busse R. Оксид азота модулирует экспрессию хемоаттрактантного белка 1 моноцитов в культивируемых эндотелиальных клетках человека. Circ Res. 1995; 76: 980–986. [PubMed] [Google Scholar] 81. Арндт Х., Смит С.В., Грейнджер Д.Н. Адгезия лейкоцитов и эндотелиальных клеток у крыс со спонтанной гипертензией и нормотензией.Гипертония. 1993; 21: 667–673. [PubMed] [Google Scholar] 82. Кубес П., Сузуки М, Грейнджер Д.Н. Оксид азота: эндогенный модулятор адгезии лейкоцитов. Proc Natl Acad Sci USA. 1991; 88: 4651–4655. [Бесплатная статья PMC] [PubMed] [Google Scholar] 83. Dimmeler S, Zeiher AM. Оксид азота — фактор выживания эндотелиальных клеток. Смерть клетки отличается. 1999; 6: 964–968. [PubMed] [Google Scholar] 84. Garg UC, Hassid A. Вазодилататоры, вырабатывающие оксид азота, и 8-бромциклический гуанозинмонофосфат ингибируют митогенез и пролиферацию культивированных клеток гладких мышц сосудов крыс.J Clin Invest. 1989; 83: 1774–1777. [Бесплатная статья PMC] [PubMed] [Google Scholar] 85. Накаки Т., Накаяма М., Като Р. Ингибирование оксидом азота и производящими оксид азота вазодилататорами синтеза ДНК в гладкомышечных клетках сосудов. Eur J Pharmacol. 1990; 189: 347–353. [PubMed] [Google Scholar] 86. Nunokawa Y, Tanaka S. Гамма-интерферон подавляет пролиферацию гладкомышечных клеток сосудов крыс за счет образования оксида азота. Biochem Biophys Res Commun. 1992; 188: 409–415. [PubMed] [Google Scholar] 87. Хоган М, Керами А, Букала Р.Конечные продукты улучшенного гликозилирования блокируют антипролиферативный эффект оксида азота. Роль в сосудистых и почечных осложнениях сахарного диабета. J Clin Invest. 1992; 90: 1110–1115. [Бесплатная статья PMC] [PubMed] [Google Scholar] 88. Саутгейт К., Ньюби АС. Индуцированная сывороткой пролиферация гладкомышечных клеток аорты кролика из сократительного состояния ингибируется 8-Br-цАМФ, но не 8-Br-цГМФ. Атеросклероз. 1990; 82: 113–123. [PubMed] [Google Scholar] 89. Förstermann U. Окислительный стресс при сосудистых заболеваниях: причины, защитные механизмы и возможные методы лечения.Nat Clin Pract Cardiovasc Med. 2008; 5: 338–349. [PubMed] [Google Scholar] 90. Хан RN, Стюарт DJ. Дефектное развитие сосудов легких у мышей с дефицитом синтазы оксида азота эндотелия. Trends Cardiovasc Med. 2006; 16: 29–34. [PubMed] [Google Scholar] 91. Мурохара Т., Асахара Т., Сильвер М., Баутерс К., Масуда Х., Калка К., Кирни М., Чен Д., Саймс Дж. Ф., Фишман М. С., Хуанг П. Л., Иснер Дж. М.. Синтаза оксида азота модулирует ангиогенез в ответ на ишемию ткани. J Clin Invest. 1998. 101: 2567–2578. [Бесплатная статья PMC] [PubMed] [Google Scholar] 92.Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Существенная роль эндотелиальной синтазы оксида азота для мобилизации стволовых клеток и клеток-предшественников. Nat Med. 2003. 9: 1370–1376. [PubMed] [Google Scholar] 93. Wohlfart P, Xu H, Endlich A, Habermeier A, Closs EI, Hubschle T, Mang C, Strobel H, Suzuki T, Kleinert H, Förstermann U, Ruetten H, Li H. Антиатеросклеротические эффекты низкомолекулярных соединений, усиливающих эндотелиальный Экспрессия синтазы оксида азота (eNOS) и предотвращение разобщения eNOS.J Pharmacol Exp Ther. 2008. 325: 370–379. [PubMed] [Google Scholar] 94. Sasaki K, Heeschen C, Aicher A, Ziebart T, Honold J, Urbich C, Rossig L, Koehl U, Koyanagi M, Mohamed A, Brandes RP, Martin H, Zeiher AM, Dimmeler S. Предварительная обработка мононуклеарных клеток костного мозга ex vivo с энхансером эндотелиальной NO-синтазы AVE9488 усиливает их функциональную активность для клеточной терапии. Proc Natl Acad Sci USA. 2006; 103: 14537–14541. [Бесплатная статья PMC] [PubMed] [Google Scholar] 95. Landmesser U, Engberding N, Bahlmann FH, Schaefer A, Wiencke A, Heineke A, Spiekermann S, Hilfiker-Kleiner D, Templin C, Kotlarz D, Mueller M, Fuchs M, Hornig B, Haller H, Drexler H.Вызванное статинами улучшение мобилизации эндотелиальных клеток-предшественников, неоваскуляризации миокарда, функции левого желудочка и выживаемости после экспериментального инфаркта миокарда требует эндотелиальной синтазы оксида азота. Тираж. 2004; 110: 1933–1939. [PubMed] [Google Scholar] 96. Чен А.Ф., Рен Дж., Мяо С.Ю. Генная терапия синтазы оксида азота при сердечно-сосудистых заболеваниях. Jpn J Pharmacol. 2002. 89: 327–336. [PubMed] [Google Scholar] 97. О’Коннор Д.М., О’Брайен Т. Генная терапия синтазой оксида азота: прогресс и перспективы.Экспертное мнение Biol Ther. 2009; 9: 867–878. [PubMed] [Google Scholar] 98. Мюллер К.Ф., Лауд К., МакНалли Дж.С., Харрисон Д.Г. Редокс-механизмы в кровеносных сосудах. Артериосклер Thromb Vasc Biol. 2005. 25: 274–278. [PubMed] [Google Scholar] 99. Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr. Генерация супероксида эндотелиальной синтазой оксида азота: влияние кофакторов. Proc Natl Acad Sci USA. 1998; 95: 9220–9225. [Бесплатная статья PMC] [PubMed] [Google Scholar] 100.Milstien S, Katusic Z. Окисление тетрагидробиоптерина пероксинитритом: последствия для функции эндотелия сосудов. Biochem Biophys Res Commun. 1999; 263: 681–684. [PubMed] [Google Scholar] 101. Ландмессер У., Дикалов С., Прайс С.Р., Макканн Л., Фукаи Т., Холланд С.М., Митч В.Е., Харрисон Д.Г. Окисление тетрагидробиоптерина приводит к разъединению синтазы оксида азота эндотелиальных клеток при гипертонии. J Clin Invest. 2003; 111: 1201–1209. [Бесплатная статья PMC] [PubMed] [Google Scholar] 102. Zou MH, Shi C, Cohen RA.Окисление цинк-тиолатного комплекса и разобщение эндотелиальной синтазы оксида азота пероксинитритом. J Clin Invest. 2002; 109: 817–826. [Бесплатная статья PMC] [PubMed] [Google Scholar] 103. Вернер Э. Р., Горрен А. С., Хеллер Р., Вернер-Фельмайер Г., Майер Б. Тетрагидробиоптерин и оксид азота: механистические и фармакологические аспекты. Exp Biol Med. 2003; 228: 1291–1302. [PubMed] [Google Scholar] 104. Heller R, Werner-Felmayer G, Werner ER. Антиоксиданты и эндотелиальный синтез оксида азота. Eur J Clin Pharmacol.2006; 62 (Приложение 13)): 21–28. [Google Scholar] 105. Вернер-Фельмайер Г., Вернер Э. Р., Фукс Д., Хаузен А., Рейбнеггер Г., Шмидт К., Вайс Г., Вахтер Х. Биосинтез птеридина в эндотелиальных клетках человека. Влияние на опосредованное оксидом азота образование циклического GMP. J Biol Chem. 1993; 268: 1842–1846. [PubMed] [Google Scholar] 106. Розенкранц-Вайс П., Сесса В.К., Милстен С., Кауфман С., Уотсон Калифорния, Побер Дж. С.. Регуляция синтеза оксида азота провоспалительными цитокинами в эндотелиальных клетках пупочной вены человека. Повышение уровня тетрагидробиоптерина увеличивает специфическую активность эндотелиальной синтазы оксида азота.J Clin Invest. 1994; 93: 2236–2243. [Бесплатная статья PMC] [PubMed] [Google Scholar] 107. Shinozaki K, Kashiwagi A, Nishio Y, Okamura T., Yoshida Y, Masada M, Toda N, Kikkawa R. Аномальный метаболизм биоптерина является основной причиной нарушения эндотелий-зависимой релаксации из-за оксида азота / O 2 — дисбаланс в инсулинорезистентная аорта крысы. Диабет. 1999; 48: 2437–2445. [PubMed] [Google Scholar] 108. Хун Х-Дж, Сяо Г, Ченг Т-Х, Йен М-Х. Прием тетрагидробиоптерина подавляет развитие гипертонии у крыс со спонтанной гипертензией.Гипертония. 2001; 38: 1044–1048. [PubMed] [Google Scholar] 109. Лаурсен Дж. Б., Сомерс М., Курц С., Макканн Л., Варнхольц А., Фриман Б. А., Тарпи М., Фукаи Т., Харрисон Д. Г.. Эндотелиальная регуляция вазомоции у мышей с дефицитом апоЕ: последствия для взаимодействий между пероксинитритом и тетрагидробиоптерином. Тираж. 2001; 103: 1282–1288. [PubMed] [Google Scholar] 110. Stroes E, Kastelein J, Cosentino F, Erkelens W, Wever R, Koomans H, Luscher T., Rabelink T. Тетрагидробиоптерин восстанавливает эндотелиальную функцию при гиперхолестеринемии.J Clin Invest. 1997; 99: 41–46. [Бесплатная статья PMC] [PubMed] [Google Scholar] 111. Heitzer T, Krohn K, Albers S, Meinertz T. Тетрагидробиоптерин улучшает эндотелий-зависимую вазодилатацию за счет увеличения активности оксида азота у пациентов с сахарным диабетом II типа. Диабетология. 2000; 43: 1435–1438. [PubMed] [Google Scholar] 112. Хигаси Ю., Сасаки С., Накагава К., Фукуда Ю., Мацуура Х, Осима Т., Чаяма К. Тетрагидробиоптерин усиливает реакцию сосудов предплечья на ацетилхолин как у нормотензивных, так и у гипертонических людей.Am J Hypertens. 2002; 15: 326–332. [PubMed] [Google Scholar] 113. Хисикава К., Накаки Т., Сузуки Х., Като Р., Сарута Т. Роль пути оксида азота l-аргинина в гипертонии. J Hypertens. 1993; 11: 639–645. [PubMed] [Google Scholar] 114. Имаидзуми Т., Хироока Ю., Масаки Х., Харада С., Момохара М., Тагава Т., Такешита А. Влияние l-аргинина на сосуды предплечья и реакции на ацетилхолин. Гипертония. 1992; 20: 511–517. [PubMed] [Google Scholar] 115. Drexler H, Zeiher AM, Meinzer K, Just H. Коррекция эндотелиальной дисфункции в коронарной микроциркуляции у пациентов с гиперхолестеринемией с помощью l-аргинина.Ланцет. 1991; 338: 1546–1550. [PubMed] [Google Scholar] 116. Россич Э., Александр Э., Блэк ПМ, Кук JP. l-аргинин нормализует функцию эндотелия сосудов головного мозга у кроликов с гиперхолестеринемией. J Clin Invest. 1991; 87: 1295–1299. [Бесплатная статья PMC] [PubMed] [Google Scholar] 117. Клосс EI, Scheld JS, Sharafi M, Förstermann U. Поставка субстрата для синтазы оксида азота в макрофагах и эндотелиальных клетках: роль переносчиков катионных аминокислот. Mol Pharmacol. 2000; 57: 68–74. [PubMed] [Google Scholar] 118.Pollock JS, Förstermann U, Mitchell JA, Warner TD, Schmidt HH, Nakane M, Murad F. Очистка и характеристика синтазы релаксирующего фактора, полученной из эндотелия, из культивируемых и природных эндотелиальных клеток аорты крупного рогатого скота. Proc Natl Acad Sci USA. 1991; 88: 10480–10484. [Бесплатная статья PMC] [PubMed] [Google Scholar] 119. Саймон А., Плис Л., Хабермайер А., Мартин Ю., Рейнинг М., Клосс Э. Роль транспорта нейтральных аминокислот и распада белка в субстрате синтазы оксида азота в эндотелиальных клетках человека.Circ Res. 2003. 93: 813–820. [PubMed] [Google Scholar] 120. Хеккер М., Сесса В.С., Харрис Х.Дж., Анггард Е.Е., Вейн-младший. Метаболизм l-аргинина и его значение для биосинтеза фактора релаксации эндотелия: культивируемые эндотелиальные клетки рециркулируют l-цитруллин в l-аргинин. Proc Natl Acad Sci USA. 1990; 87: 8612–8616. [Бесплатная статья PMC] [PubMed] [Google Scholar] 121. Bivalacqua TJ, Hellstrom WJ, Kadowitz PJ, Чемпион ХК. Повышенная экспрессия аргиназы II в пещеристых телах человека с диабетом: при эректильной дисфункции, связанной с диабетом.Biochem Biophys Res Commun. 2001; 283: 923–927. [PubMed] [Google Scholar] 122. Сюй В., Канеко FT, Чжэн С., Комхейр С.А., Яноча А.Дж., Гогганс Т., Тунниссен Ф.Б., Фарвер С., Хазен С.Л., Дженнингс С., Дваик Р.А., Арролига А.С., Эрзурум СК. Повышение аргиназы II и снижение синтеза NO в эндотелиальных клетках пациентов с легочной артериальной гипертензией. FASEB J. 2004; 18: 1746–1748. [PubMed] [Google Scholar] 123. Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, Burke S, Shoukas AA, Nyhan D, Champion HC, Hare JM.Аргиназа реципрокно регулирует активность синтазы оксида азота и способствует эндотелиальной дисфункции в стареющих кровеносных сосудах. Тираж. 2003; 108: 2000–2006. [PubMed] [Google Scholar] 124. Ming XF, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L, Hayoz D, Ruffieux J, Rusconi S., Montani JP, Yang Z. Тромбин стимулирует ферментативную активность эндотелиальной аргиназы человека через путь RhoA / ROCK: последствия для атеросклеротического эндотелия дисфункция. Тираж. 2004. 110: 3708–3714. [PubMed] [Google Scholar] 125.Gorren AC, List BM, Schrammel A, Pitters E, Hemmens B, Werner ER, Schmidt K, Mayer B. Не содержащая тетрагидробиоптерина нейрональная синтаза оксида азота: данные о двух идентичных высоко антикооперативных сайтах связывания птеридина. Биохимия. 1996; 35: 16735–16745. [PubMed] [Google Scholar] 126. Martasek P, Miller RT, Liu Q, Roman LJ, Salerno JC, Migita CT, Raman CS, Gross SS, Ikeda-Saito M, Masters BS. Мутант C331A нейрональной синтазы оксида азота нарушает связывание аргинина. J Biol Chem. 1998; 273: 34799–34805.[PubMed] [Google Scholar] 127. Sydow K, Munzel T. ADMA и окислительный стресс. Atheroscler Suppl. 2003; 4: 41–51. [PubMed] [Google Scholar] 128. Богер Р. Х., Салливан Л. М., Шведхельм Е., Ван Т. Дж., Маас Р., Бенджамин Е. Дж., Шульце Ф., Ксантакис В., Бенндорф Р. А., Васан Р. С.. Асимметричный диметиларгинин в плазме и частота сердечно-сосудистых заболеваний и смерти среди населения. Тираж. 2009; 119: 1592–1600. [Бесплатная статья PMC] [PubMed] [Google Scholar] 129. Антониадис С., Широдария С., Лисон П., Антонопулос А., Уоррик Н., Ван-Аше Т., Каннингтон К., Тусулис Д., Пиллаи Р., Ратнатунга С., Стефанадис К., Чаннон К.М.Связь асимметричного диметиларгинина в плазме (ADMA) с повышенным производством супероксида в сосудах и разобщением эндотелиальной синтазы оксида азота: последствия для эндотелиальной функции при атеросклерозе человека. Eur Heart J. 2009; 30: 1142–1150. [PubMed] [Google Scholar] 130. Линь К.Ю., Ито А., Асагами Т., Цао П.С., Адимоолам С., Кимото М., Цудзи Х., Ривен Г.М., Кук Дж.П. Нарушение пути синтазы оксида азота при сахарном диабете: роль асимметричного диметиларгинина и диметиларгининдиметиламиногидролазы.Тираж. 2002; 106: 987–992. [PubMed] [Google Scholar] 131. Böger RH, Sydow K, Borlak J, Thum T, Lenzen H, Schubert B, Tsikas D, Bode-Böger SM. Холестерин ЛПНП активирует синтез асимметричного диметиларгинина в эндотелиальных клетках человека: участие S-аденозилметионин-зависимых метилтрансфераз. Circ Res. 2000. 87: 99–105. [PubMed] [Google Scholar] 132. Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. S-глутатионилирование разъединяет eNOS и регулирует его клеточную и сосудистую функцию.Природа. 2010; 468: 1115–1118. [Бесплатная статья PMC] [PubMed] [Google Scholar] 133. Zweier J, Chen CA, Druhan LJ. S-глутатионилирование меняет наше понимание разобщения eNOS и передачи сигналов, опосредованной NO / ROS. Антиоксидный окислительно-восстановительный сигнал. 2011; 14: 1769–1775. [Бесплатная статья PMC] [PubMed] [Google Scholar] 134. Иманиши Т., Икедзима Х, Цудзиока Х, Курои А., Кобаяши К., Мурагаки Ю., Мочизуки С., Гото М., Йошида К., Акасака Т. Добавление эплеренона к ингибитору ангиотензин-превращающего фермента эффективно улучшает биодоступность оксида азота.Гипертония. 2008; 51: 734–741. [PubMed] [Google Scholar] 135. Griendling KK, Sorescu D, Ushio-Fukai M. NAD (P) H оксидаза: роль в сердечно-сосудистой биологии и болезнях. Circ Res. 2000; 86: 494–501. [PubMed] [Google Scholar] 136. Nickenig G, Baumer AT, Temur Y, Kebben D, Jockenhovel F, Bohm M. Статин-чувствительное нарушение регуляции функции и плотности рецептора AT1 у мужчин с гиперхолестеринемией. Тираж. 1999; 100: 2131–2134. [PubMed] [Google Scholar] 137. Иманиши Т, Цудзиока Х, Икедзима Х, Курои А, Такарада С., Китабата Х, Танимото Т, Мурагаки Й, Мотидзуки С., Гото М, Йошида К., Акасака Т.Ингибитор ренина алискирен улучшает нарушенную биодоступность оксида азота и защищает от атеросклеротических изменений. Гипертония. 2008. 52: 563–572. [PubMed] [Google Scholar] 138. Klingbeil AU, John S, Schneider MP, Jacobi J, Handrock R, Schmieder RE. Влияние блокады рецептора AT1 на функцию эндотелия при гипертонической болезни. Am J Hypertens. 2003. 16: 123–128. [PubMed] [Google Scholar] 139. Wassmann S, Hilgers S, Laufs U, Bohm M, Nickenig G. Антагонизм рецептора 1 типа ангиотензина II улучшает эндотелиальную дисфункцию, связанную с гиперхолестеринемией.Артериосклер Thromb Vasc Biol. 2002; 22: 1208–1212. [PubMed] [Google Scholar] 140. Warnholtz A, Nickenig G, Schulz E, Macharzina R, Brasen JH, Skatchkov M, Heitzer T., Stasch JP, Griendling KK, Harrison DG, Bohm M, Meinertz T, Munzel T. стадии атеросклероза: данные о вовлечении ренин-ангиотензиновой системы. Тираж. 1999; 99: 2027–2033. [PubMed] [Google Scholar] 141. Mancini GB, Henry GC, Macaya C, O’Neill BJ, Pucillo AL, Carere RG, Wargovich TJ, Mudra H, Luscher TF, Klibaner MI, Haber HE, Uprichard AC, Pepine CJ, Pitt B.Ингибирование ангиотензин-превращающего фермента квинаприлом улучшает эндотелиальную вазомоторную дисфункцию у пациентов с ишемической болезнью сердца. Исследование ТРЕНД (Испытание по обращению Эндотелиальной дисфункции). Тираж. 1996. 94: 258–265. [PubMed] [Google Scholar] 142. Hornig B, Landmesser U, Kohler C, Ahlersmann D, Spiekermann S, Christoph A, Tatge H, Drexler H. Сравнительный эффект ингибирования ace и антагонизма рецептора ангиотензина II типа 1 на биодоступность оксида азота у пациентов с ишемической болезнью сердца: роль супероксиддисмутаза.Тираж. 2001; 103: 799–805. [PubMed] [Google Scholar] 143. Bauersachs J, Fraccarollo D. Больше NO — больше никаких АФК: комбинированная селективная блокада минералокортикоидных рецепторов и ингибирование ангиотензин-превращающего фермента для защиты сосудов. Гипертония. 2008. 51: 624–625. [PubMed] [Google Scholar] 144. Сато М., Фудзимото С., Аракава С., Яда Т., Намикоши Т., Харуна Ю., Хорике Н., Сасаки Т., Кашихара Н. Блокатор рецепторов ангиотензина II типа 1 улучшает несвязанную эндотелиальную синтазу оксида азота у крыс с экспериментальной диабетической нефропатией.Пересадка нефрола Dial. 2008. 23: 3806–3813. [Бесплатная статья PMC] [PubMed] [Google Scholar] 145. Liao JK. Помимо снижения уровня липидов: роль статинов в защите сосудов. Int J Cardiol. 2002; 86: 5–18. [PubMed] [Google Scholar] 147. John S, Schlaich M, Langenfeld M, Weihprecht H, Schmitz G, Weidinger G, Schmieder RE. Повышенная биодоступность оксида азота после гиполипидемической терапии у пациентов с гиперхолестеринемией: рандомизированное плацебо-контролируемое двойное слепое исследование. Тираж. 1998. 98: 211–216.[PubMed] [Google Scholar] 148. Laufs U, Liao JK. Посттранскрипционная регуляция стабильности мРНК эндотелиальной синтазы оксида азота с помощью Rho GTPase. J Biol Chem. 1998; 273: 24266–24271. [PubMed] [Google Scholar] 149. Ферон О, Десси С., Десагер Дж. П., Баллиганд Дж. Л. Ингибирование гидроксиметилглутарил-коэнзим А-редуктазы способствует активации эндотелиальной синтазы оксида азота за счет снижения содержания кавеолина. Тираж. 2001. 103: 113–118. [PubMed] [Google Scholar] 150. Курейши Ю., Ло З., Сиодзима И., Бялик А., Фултон Д., Лефер Д. Д., Сесса В. К., Уолш К.Ингибитор HMG-CoA редуктазы симвастатин активирует протеинкиназу Akt и способствует ангиогенезу у нормохолестеринемических животных. Nat Med. 2000; 6: 1004–1010. [Бесплатная статья PMC] [PubMed] [Google Scholar] 151. Wagner AH, Kohler T, Ruckschloss U, Just I, Hecker M. Улучшение зависимого от оксида азота вазодилатации ингибиторами HMG-CoA редуктазы за счет ослабления образования эндотелиального супероксид-аниона. Артериосклер Thromb Vasc Biol. 2000. 20: 61–69. [PubMed] [Google Scholar] 152. Хаттори Ю., Наканиси Н., Акимото К., Ёсида М., Касаи К.Ингибитор HMG-CoA редуктазы увеличивает мРНК GTP циклогидролазы I и тетрагидробиоптерин в эндотелиальных клетках сосудов. Артериосклер Thromb Vasc Biol. 2003. 23: 176–182. [PubMed] [Google Scholar] 153. Венцель П., Дайбер А., Эльце М., Брандт М., Клосс Е., Сюй Дж., Тум Т., Бауэрсакс Дж., Эртл Г., Зоу М. Х., Фёрстерманн Ю., Мюнцель Т. Механизмы, лежащие в основе воссоединения eNOS с помощью ингибирования HMG-CoA редуктазы у крыс модель сахарного диабета, индуцированного стрептозотоцином. Атеросклероз. 2008; 198: 65–76. [Бесплатная статья PMC] [PubMed] [Google Scholar] 154.Ниссен С.Е., Николлс С.Дж., Сипахи И., Либби П., Райхлен Дж.С., Баллантайн К.М., Давиньон Дж., Эрбель Р., Фрукарт Дж. К., Тардиф Дж. К., Шенхаген П., Кроу Т., Каин В., Вольски К., Гурмастик М., Тузку Е. М.. Эффект очень высокоинтенсивной терапии статинами на регресс коронарного атеросклероза: исследование ASTEROID. ДЖАМА. 2006; 295: 1556–1565. [PubMed] [Google Scholar] 155. Пател Т.Н., Шишехбор М.Х., Бхатт Д.Л. Обзор терапии высокими дозами статинов: воздействие на холестерин и воспаление при атеросклерозе. Eur Heart J. 2007; 28: 664–672.[PubMed] [Google Scholar] 156. Николсон С., Бонечини-Алмейда Мда Дж., Лапа и Сильва Дж. Р., Натан С., Се К. В., Мамфорд Р., Вайднер Дж. Индуцируемая синтаза оксида азота в легочных альвеолярных макрофагах больных туберкулезом. J Exp Med. 1996; 183: 2293–2302. [Бесплатная статья PMC] [PubMed] [Google Scholar] 157. MacMicking J, Xie QW, Nathan C. Оксид азота и функция макрофагов. Анну Рев Иммунол. 1997; 15: 323–350. [PubMed] [Google Scholar] 158. Стенджер С., Тюринг Х., Роллингхофф М., Богдан К.Тканевая экспрессия индуцибельной синтазы оксида азота тесно связана с устойчивостью к Leishmania major . J Exp Med. 1994; 180: 783–793. [Бесплатная статья PMC] [PubMed] [Google Scholar] 159. Вэй XQ, Чарльз И.Г., Смит А., Уре Дж., Фэн Дж. Дж., Хуанг Ф. П., Сюй Д., Мюллер В., Монкада С., Лью Ф.Й. Измененные иммунные ответы у мышей, лишенных индуцибельной синтазы оксида азота. Природа. 1995; 375: 408–411. [PubMed] [Google Scholar] 160. Рафи П., Огава Х., Хайдеманн Дж., Ли М.С., Аслам М., Ламиранд Т.Х., Фишер П.Дж., Грэвин С.Дж., Двинелл М.Б., Джонсон С.П., Шейкер Р., Бинион Д.Г.Выделение и характеристика эндотелиальных клеток микрососудов пищевода человека: механизмы воспалительной активации. Am J Physiol Gastrointest Liver Physiol. 2003; 285: G1277 – G1292. [PubMed] [Google Scholar] 161. Вонг Дж. М., Биллиар Т. Р.. Регуляция и функция индуцибельной синтазы оксида азота при сепсисе и остром воспалении. Adv Pharmacol. 1995; 34: 155–170. [PubMed] [Google Scholar] 162. Ли Х., Фёрстерманн У. Оксид азота в патогенезе сосудистых заболеваний. J Pathol. 2000; 190: 244–254. [PubMed] [Google Scholar] 163.Klatt P, Pfeiffer S, List BM, Lehner D, Glatter O, Bachinger HP, Werner ER, Schmidt K, Mayer B. Характеристика гемодефицитной нейрональной синтазы оксида азота показывает роль гема в димеризации субъединиц и связывании аминогруппы. кислотный субстрат и тетрагидробиоптерин. J Biol Chem. 1996; 271: 7336–7342. [PubMed] [Google Scholar] 164. Коцонис П., Фрелих Л.Г., Шутенко З.В., Хорейси Р., Пфлейдерер В., Шмидт Х. Х. Аллостерическая регуляция нейрональной синтазы оксида азота тетрагидробиоптерином и подавление самоповреждающего супероксида.Biochem J. 2000; 346 (Pt 3): 767–776. [Бесплатная статья PMC] [PubMed] [Google Scholar] 165. Abu-Soud HM, Stuehr DJ. Синтазы оксида азота показывают роль кальмодулина в контроле переноса электронов. Proc Natl Acad Sci USA. 1993; 90: 10769–10772. [Бесплатная статья PMC] [PubMed] [Google Scholar] 166. List BM, Klosch B, Volker C, Gorren AC, Sessa WC, Werner ER, Kukovetz WR, Schmidt K, Mayer B. Характеристика бычьей эндотелиальной синтазы оксида азота как гомодимера с подавленной несвязанной активностью NADPH-оксидазы: кинетика связывания тетрагидробиоптерина и роль гема в димеризации.Biochem J. 1997; 323 (Pt 1): 159–165. [Бесплатная статья PMC] [PubMed] [Google Scholar] 167. Бруннер К., Торчанов А., Хемменс Б., Эндрю П.Дж., Майер Б., Кунгл А.Дж. Чувствительность динамики флуоресценции флавинов в нейрональной синтазе оксида азота к кофактор-индуцированным конформационным изменениям и димеризации. Биохимия. 1998; 37: 17545–17553. [PubMed] [Google Scholar] 168. Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Förstermann U, Meinertz T, Griendling K, Munzel T.Влияние инфузии ангиотензина II на экспрессию и функцию NAD (P) H оксидазы и компонентов передачи сигналов оксида азота / цГМФ. Circ Res. 2002; 90: E58 – E65. [PubMed] [Google Scholar] 169. Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, Meinertz T, Griendling K, Harrison DG, Förstermann U, Münzel T. Механизмы, лежащие в основе эндотелиальной дисфункции в сахарный диабет. Circ Res. 2001; 88: E14 – E22. [PubMed] [Google Scholar] 170. Драммонд Г. Р., Кай Х., Дэвис М. Е., Рамасами С., Харрисон Д. Г..Транскрипционная и посттранскрипционная регуляция экспрессии эндотелиальной синтазы оксида азота перекисью водорода. Circ Res. 2000. 86: 347–354. [PubMed] [Google Scholar] 171. Li H, Oehrlein SA, Wallerath T, Ihrig-Biedert I, Wohlfart P, Ulshöfer T, Jessen T, Herget T, Förstermann U, Kleinert H. Активация протеинкиназы C альфа и / или эпсилон усиливает транскрипцию эндотелиального оксида азота человека ген синтазы. Mol Pharmacol. 1998. 53: 630–637. [PubMed] [Google Scholar] 172. Горрен А.С., Кунгл А.Дж., Шмидт К., Вернер Э.Р., Майер Б.Электрохимия кофакторов птерина и ингибиторов синтазы оксида азота. Оксид азота. 2001. 5: 176–186. [PubMed] [Google Scholar] 173. Бек Н., Горрен А.Ф., Майер Б., Шмидт П.П., Андерссон К.К., Ланге Р. Роль тетрагидробиоптерина в активации кислорода синтазой оксида азота. J Inorg Biochem. 2000. 81: 207–211. [PubMed] [Google Scholar]Произошла ошибка при настройке пользовательского файла cookie
Этот сайт использует файлы cookie для повышения производительности. Если ваш браузер не принимает файлы cookie, вы не можете просматривать этот сайт.
Настройка вашего браузера для приема файлов cookie
Существует множество причин, по которым cookie не может быть установлен правильно. Ниже приведены наиболее частые причины:
- В вашем браузере отключены файлы cookie. Вам необходимо сбросить настройки вашего браузера, чтобы он принимал файлы cookie, или чтобы спросить вас, хотите ли вы принимать файлы cookie.
- Ваш браузер спрашивает вас, хотите ли вы принимать файлы cookie, и вы отказались. Чтобы принять файлы cookie с этого сайта, нажмите кнопку «Назад» и примите файлы cookie.
- Ваш браузер не поддерживает файлы cookie. Если вы подозреваете это, попробуйте другой браузер.
- Дата на вашем компьютере в прошлом. Если часы вашего компьютера показывают дату до 1 января 1970 г., браузер автоматически забудет файл cookie. Чтобы исправить это, установите правильное время и дату на своем компьютере.
- Вы установили приложение, которое отслеживает или блокирует установку файлов cookie. Вы должны отключить приложение при входе в систему или проконсультироваться с системным администратором.
Почему этому сайту требуются файлы cookie?
Этот сайт использует файлы cookie для повышения производительности, запоминая, что вы вошли в систему, когда переходите со страницы на страницу. Чтобы предоставить доступ без файлов cookie потребует, чтобы сайт создавал новый сеанс для каждой посещаемой страницы, что замедляет работу системы до неприемлемого уровня.
Что сохраняется в файле cookie?
Этот сайт не хранит ничего, кроме автоматически сгенерированного идентификатора сеанса в cookie; никакая другая информация не фиксируется.
Как правило, в cookie-файлах может храниться только информация, которую вы предоставляете, или выбор, который вы делаете при посещении веб-сайта. Например, сайт не может определить ваше имя электронной почты, пока вы не введете его. Разрешение веб-сайту создавать файлы cookie не дает этому или любому другому сайту доступа к остальной части вашего компьютера, и только сайт, который создал файл cookie, может его прочитать.
Прямая активация эндотелиальной синтазы оксида азота обеспечивает атерозащиту при атеросклерозе, ускоренном диабетом
Abstract
Пациенты с диабетом имеют повышенный риск развития атеросклероза.Эндотелиальная дисфункция, характеризующаяся пониженной биодоступностью NO-производной эндотелиальной NO-синтазы (eNOS), является критическим индуктором атеросклероза. Однако защитный аспект eNOS при атеросклерозе, ассоциированном с диабетом, остается спорным, что, вероятно, является следствием его способности выделять как защитный NO, так и вредные кислородные радикалы в нормальных условиях и при болезни, соответственно. Использование атеропротекторной активности eNOS в условиях диабета остается труднодостижимым, отчасти из-за отсутствия эндогенных активаторов высвобождения NO, специфичных для eNOS.Недавно мы показали in vitro, что высвобождение NO из eNOS может быть увеличено путем блокирования его связывания с кавеолином-1, основным белком оболочки кавеол, с помощью высокоспецифичного пептида CavNOxin. Однако неизвестно, может ли нацеливание на eNOS с использованием этого пептида ослабить атеросклероз, связанный с диабетом. В этом исследовании мы показываем, что CavNOxin может уменьшить атеросклеротическую нагрузку на ~ 84% in vivo. Напротив, мыши, лишенные eNOS, проявляют устойчивость к лечению CavNOxin, что указывает на специфичность eNOS. Механически CavNOxin снижает маркеры окислительного стресса, ингибирует экспрессию проатерогенных медиаторов и блокирует лейкоцитарно-эндотелиальные взаимодействия.Эти данные впервые показывают, что активация эндогенной eNOS может обеспечивать атерозащиту при диабете, и позволяют предположить, что CavNOxin является жизнеспособной стратегией для разработки антиатеросклеротических соединений.
Введение
Диабет широко считается независимым фактором риска развития сердечно-сосудистых заболеваний, при этом около 80% смертности и заболеваемости от сердечно-сосудистых заболеваний связаны с макрососудистыми осложнениями, такими как атеросклероз (1–3). Повышенный риск развития сосудистых осложнений у лиц с диабетом типа 1 (T1D) или типа 2 (T2D) возникает, несмотря на интенсивный гликемический контроль, что подчеркивает необходимость новых подходов для уменьшения бремени макроваскулярных повреждений, опосредованных диабетом (4).
Эндотелий сосудов играет решающую роль в диабете, связанном с атеросклерозом, регулируя проницаемость сосудов, воспаление, координацию движения лейкоцитов и тромбоз (1,5). Действительно, функция эндотелия сосудов значительно нарушается во время диабета, это явление называется эндотелиальной дисфункцией и характеризуется сниженной биодоступностью важного медиатора эндотелиальных клеток, оксида азота (NO). Такое хроническое ослабление высвобождения NO из эндотелия способствует активации и адгезии тромбоцитов и лейкоцитов, нарушает целостность барьера эндотелиальных клеток и вызывает активацию провоспалительных генов (6-8).Более того, сниженная NO-зависимая вазодилатация и повышенная адгезия лейкоцитов к эндотелию, оба из которых являются отличительными признаками эндотелиальной дисфункции, наблюдались у пациентов с диабетом и диабетических животных на моделях (9–12). Точно так же в культивируемых эндотелиальных клетках, подвергшихся воздействию высокого уровня глюкозы, наблюдалось пониженное высвобождение NO, вызванное эндотелиальной NO-синтазой (eNOS) (13-15). Кроме того, эндотелиальная дисфункция, диабет и атеросклероз связаны с повышенным состоянием окислительного стресса и его основным медиатором, супероксид-анионом.Супероксид может улавливать свободный NO с образованием пероксинитрита, еще одной мощной реактивной формы кислорода (АФК), которая в значительной степени способствует снижению биодоступности NO.
Атеропротекторный NO высвобождается эндотелием через конститутивно экспрессируемую eNOS, димерную изоформу NOS, которая активируется гемодинамическими факторами и агонистами. Генетическая делеция или фармакологическое ингибирование eNOS в доклинических моделях значительно увеличивает атеросклеротическую нагрузку, обеспечивая четкие доказательства антиатерогенной роли NO, производного от eNOS (16,17).Однако сверхэкспрессия eNOS у мышей также ускоряет атеросклероз, что, вероятно, является следствием разобщения фермента до его мономерного состояния, что вместо этого приводит к образованию вредного супероксида (18). Следовательно, правильная регуляция eNOS имеет решающее значение для ее атеропротекторной функции (19). NO, происходящий из eNOS, регулируется фосфорилированием фермента и доступностью его субстратов (L-аргинин) и кофакторов (Bh5). Кроме того, для оптимального высвобождения NO требуется доставка eNOS к кавеолам, которые представляют собой обогащенные липидами органеллы на плазматической мембране, которые, как известно, поддерживают пространственную организацию сигнальных комплексов и участвуют в событиях передачи сигнала (20).Внутри кавеол базальная активность eNOS поддерживается под жестким ингибирующим контролем кавеолином-1 (Cav-1), основным белком оболочки кавеол, посредством прямого белок-белкового взаимодействия между каркасным доменом (аминокислоты 82–101) Cav- 1 и eNOS (21–24). При увеличении внутриклеточного Ca 2+ , индуцированном агонистом (фактор роста эндотелия сосудов ацетилхолина), eNOS привлекает кальмодулин и HSP90, что способствует диссоциации Cav-1 от eNOS, что приводит к взаимодействию стимула-ответ eNOS, что приводит к образованию из NO (20).Мы показали, что треонин 90,91 и особенно фенилаланин 92 (T90,91, F92) в каркасном домене Cav-1 ответственны за ингибирование высвобождения NO, происходящего из eNOS (21). Кроме того, этот ингибирующий домен отличается от связывающего домена, поскольку мутированный белок F92A Cav-1 связывает eNOS, но не может ингибировать его активность (25,26). Однако при коэкспрессии в эндотелиальных клетках, экспрессирующих eNOS и ингибирующий Cav-1, F92A Cav-1 вызывал неожиданное, но устойчивое увеличение нестимулированного высвобождения NO, происходящего из eNOS (25).Эти наблюдения выявили сильное ингибирующее действие Cav-1 на eNOS и укрепили концепцию антагонизма eNOS / Cav-1: предотвращение связывания eNOS с ингибирующим Cav-1 приводит к увеличению базального высвобождения NO. Действительно, проницаемый для клеток пептид, производный Cav-1 с T90,91, F92, замещенным на аланины, известный как CavNOxin, может увеличивать активность eNOS в реконструированных и эндотелиальных клетках (21,25) и снижать тонус сосудов ex vivo, эффект это было полностью отменено у мышей с нокаутом eNOS и Cav-1 (KO) (25).Эти высокоспецифичные для eNOS и Cav-1 биологические функции CavNOxin являются первыми, демонстрирующими повышенное высвобождение NO посредством регулируемых взаимодействий eNOS / Cav-1 при сохранении зависимых от Cav-1 биологических активностей, таких как образование кавеол (25).
В этом исследовании мы исследовали, может ли улучшение высвобождения NO за счет использования CavNOxin ослабить атеросклероз в условиях гипергликемии. Мы сообщаем, что лечение мышей с диабетом с помощью CavNOxin нормализует ряд окислительного стресса и воспалительных маркеров и ослабляет индуцированный диабетом атеросклероз до 84%, тогда как инактивация гена eNOS делает животных с диабетом устойчивыми к лечению CavNOxin.Что наиболее важно, наши данные предполагают, что экзогенные агенты могут непосредственно и специфически запускать защитную функцию eNOS, указывая на то, что это взаимодействие следует рассматривать как прямую фармакологическую мишень для уменьшения бремени диабетического повреждения органов-мишеней, таких как атеросклероз.
Дизайн и методы исследования
Животные
Аполипопротеин E (ApoE) мышей KO были приобретены в Центре ресурсов животных (Западная Австралия) и в лаборатории Джексона (Бар-Харбор, штат Мэн) и размещены в Центре медицинских исследований и образования Альфреда. объект и объект генетической инженерии в Исследовательском центре Джеймса Хогга Университета Британской Колумбии, соответственно.Все эксперименты были одобрены соответствующими комитетами по уходу за животными. Для создания мышей eNOS / ApoE double KO (dKO) мышей eNOS KO были приобретены в лаборатории Джексона и подвергнуты повторному кесареву сечению, которые использовались для создания мышей eNOS KO на чистом фоне C57BL / 6. Мышей eNOS KO скрещивали с мышами ApoE KO, и полученные мыши, гетерозиготные как по eNOS, так и по ApoE, затем скрещивали с мышами ApoE KO. Наконец, однопометники, которые были гетерозиготными по ApoE KO / eNOS, были скрещены вместе с получением одинаковых по возрасту однопометников, которые были ApoE KO и eNOS дикого типа, eNOS гетерозиготными или eNOS KO.
Схема эксперимента
Для создания инсулино-дефицитной модели, отражающей СД1, самцам мышей ApoE KO в возрасте 8 недель сделали диабет внутрибрюшинными инъекциями стрептозотоцина (STZ; 100 мг / кг / день) или цитратного буфера (недиабетический [ND ] контролирует) 2 дня подряд. Через 2 недели мышей рандомизировали и вводили внутрибрюшинно каждые 3 дня стерильную воду, пептид-носитель (Antennapedia, растворенный в 1% ДМСО в стерильной воде, та же молярная доза, что и CavNOxin) или CavNOxin в дозе 2.5 и 5,0 мг / кг (растворенный в 1% ДМСО в стерильной воде) в течение 14 недель. Предыдущие исследования нашей группы продемонстрировали, что этот период времени позволяет развиваться прочным атеросклеротическим бляшкам внутри стенок сосудов и синуса аорты у мышей с диабетом ApoE KO (27). Каждую неделю измеряли уровень глюкозы в крови без голодания, чтобы подтвердить, что у мышей был диабет на протяжении всего исследования.
Для создания инсулинорезистентной модели, отражающей T2D, самцов мышей ApoE KO в возрасте 8 недель и мышей eNOS / ApoE dKO кормили западной диетой (WD), содержащей 22% жира и 0.15% холестерина (специальные корма, SF00–219 или Harlan Teklad TD88137) ad libitum в течение 12 недель (28). Мыши ApoE KO, получавшие WD, проявляют метаболический фенотип, соответствующий T2D, который включает повышенные уровни инсулина и глюкозы по сравнению с мышами, получавшими стандартную пищу (28-30). В течение 12-недельного периода времени мышам вводили внутрибрюшинно каждые 3 дня либо пептид-носитель, либо CavNOxin (2,5 мг / кг). Контрольную группу мышей-самцов ApoE KO кормили стандартной пищей и подвергали той же схеме инъекций, что и мышей ApoE KO, получавших WD.
Анализ атеросклеротических поражений
В конечной точке исследования мышей умерщвляли и атеросклеротические поражения оценивали, как описано ранее, с использованием метода анфас после окрашивания Суданом-IV для оценки поражений в трех областях аорты (31,32). Поражения также определялись в пазухах сердца после окрашивания Oil Red O, как описано ранее (31,32). Для анализа на лицо поражения выражаются как процент бляшек, обнаруженных в определенной области, в то время как для синуса аорты размер поражения (мкм 2 ) определялся из среднего значения пяти поперечных сечений, и результаты каждой группы выражались. как размер поражения (мкм 2 ) ± SEM.
Анализ плазмы и мочи
За неделю до прекращения мышей содержали в метаболических клетках в течение 24 часов для сбора мочи. 8-изопростаны в моче определяли как маркер окислительного стресса в соответствии с инструкциями производителя (Caymen Chemical). По окончании животным не давали голодать в течение 12 часов, а после анестезии кровь (0,8–1 мл) собирали в гепарине и немедленно центрифугировали при 4000 об / мин в течение 10 минут для отделения плазмы для аналитических целей.NO в плазме определяли с использованием набора для определения общего NO, основанного на принципах реакции Грисса в соответствии с инструкциями производителя (Enzo Life Sciences). Производные реактивных метаболитов кислорода (dROM) были измерены в плазме как показатель окислительного стресса с использованием системы FRAS-4, как описано ранее (33). Глюкозу в плазме, холестерин, ЛПВП, триглицериды и инсулин измеряли с использованием коммерчески доступных ферментативных наборов, а ЛПНП определяли с использованием расчета Фридевальда.
Количественный анализ RT-PCR
В другой когорте мышей аорты мгновенно замораживали в TRIzol, и после гомогенизации ткани экстрагировали общую РНК. Без ДНКазы синтез РНК и кДНК проводили, как описано ранее (31). Зонды и праймеры были приобретены в Applied Biosystems (Фостер-Сити, Калифорния). Экспрессия гена eNOS, молекулы адгезии сосудистых клеток-1 (VCAM-1), молекулы внутриклеточной адгезии-1, MCP-1 и субъединицы p65 ядерного фактора-κB анализировалась с помощью количественной ОТ-ПЦР (qRT-PCR), как описано ранее ( 27).
Иммуногистохимия
Парафиновые срезы аорты окрашивали на нитротирозин (NT) (маркер повреждения, вызванного пероксинитритом) и 4-HNE (маркер перекисного окисления липидов), а замороженные срезы аорты окрашивали на VCAM-1. Вкратце, парафиновые срезы депарафинизировали, замороженные срезы аорты фиксировали холодным ацетоном, а эндогенные пероксидазы инактивировали 3% H 2 O 2 в трис-буферном физиологическом растворе. Срезы инкубировали с агентом, блокирующим белок, и набором, блокирующим биотин-авидин (Vector Laboratories).Для окрашивания 4-HNE в дополнение к локализации мышиных антител на тканях мыши использовали набор Mouse On Mouse (M.O.M Kit; Vector Laboratories). Затем срезы аорты инкубировали с соответствующим первичным антителом (VCAM-1: 1: 100; 4-HNE: 1: 100; и NT: 1:75) в течение ночи при 4 ° C. Затем добавляли вторичные антитела, биотинилированный иммуноглобулин против кроликов 1: 100 (Dako) или биотинилированный иммуноглобулин против крыс (1: 200; Vector Laboratories) на 30 мин, а затем стрептавидин, конъюгированный с пероксидазой хрена, разведенный 1: 500 (Dako). и инкубировали в течение 30 минут в тетрагидрохлориде 3,3′-диаминобензидина (Sigma-Aldrich) с контрастным красителем гематоксилином.Изображения визуализировали под световой микроскопией и количественно оценивали с помощью Image Pro Plus. В среднем на каждую мышь оценивали от трех до пяти срезов, а на группу — от семи до девяти мышей.
Эксперименты in vitro на эндотелиальных клетках аорты человека
Эндотелиальные клетки аорты человека (НАЕС) (Cell Application, San Diego, CA; пассаж 4-8) поддерживали в среде эндотелиальных клеток (EGM-2 bullet kit; Lonza) и засевали в экспериментальные шестилуночные чашки для культивирования, предварительно покрытые фибриногеном (100 мкг / мл).При 90% -ной конфлюэнтности HAEC голодали по сыворотке и обрабатывали либо носителем, либо пептидом CavNOxin (5 мкмоль) в течение 6 часов. Ранее мы продемонстрировали, что эта концентрация и момент времени значительно увеличивают высвобождение NO в эндотелиальных клетках и снижают ассоциацию эндогенного Cav-1 с eNOS (25). В отдельном эксперименте HAEC выращивали в среде с низким содержанием глюкозы (5 ммоль) и высоким содержанием глюкозы (30 ммоль) в течение 3 дней до обработки пептидом.
Оценка взаимодействий моноцитов / лейкоцитов-эндотелия In vitro и Ex Vivo
Взаимодействия моноцитов / эндотелия определяли путем проведения статических анализов адгезии клеток in vitro.Вкратце, человеческие моноцитарные клетки THP-1 метили с помощью набора для флуоресцентной маркировки CellVue Burgundy (Affymetrix) в соответствии с инструкциями производителя и инкубировали с обработанными HAEC (как описано выше) в течение 20 минут при 37 ° C. После этого HAEC дважды промывали PBS и фиксировали 10% нейтральным забуференным формалином, и планшеты сканировали с использованием инфракрасного сканера Odyssey и определяли количественно.
Лейкоцитарно-эндотелиальные взаимодействия определяли ex vivo путем выполнения анализов адгезии динамического потока, как описано ранее (34).Вкратце, аорты мышей ND и диабетических мышей ApoE KO инкубировали с носителем или пептидом CavNOxin (10 мкмоль; 12 часов) в присутствии или в отсутствие фактора некроза опухоли-α (TNF-α) (5 нг / мл; 4 часа). ранее нами было продемонстрировано, что оно значительно снижает вызванное фенилэфрином сокращение изолированной аорты (25). Затем сосуды осторожно устанавливали на каждом конце канюли в камере для сосудов, содержащей предварительно нагретый буфер Кребса. Цельную кровь человека (5 мл / сосуд) метили VybrantDil (1: 1000, 10 мин) и затем перфузировали через сосуд со скоростью 100 мкл / мин.Взаимодействие меченых лейкоцитов визуализировали и количественно оценивали с помощью флуоресцентного микроскопа. Показания снимали на двух полях вдоль перфузируемого сосуда в течение 10 с в 5-, 10- и 15-минутные моменты времени.
Измерение супероксида в аортах
Содержание супероксида измеряли с помощью окрашивания дигидроэтидием (DHE; 10 мкмоль / л) на замороженных срезах аорты. Вкратце, последовательные срезы мгновенно замороженной аорты делали криосрезы толщиной 10 мкм и помещали на предметные стекла (Menzel-Glaser SuperFrost Plus).Для каждой аорты окрашивание DHE проводили в присутствии или в отсутствие 1 ммоль темпола (Fluka), миметика супероксиддисмутазы, что позволяет измерять супероксид-специфичный DHE с использованием конфокального микроскопа Zeiss 510 Meta, оснащенного криптоновым / аргоновым лазером (возбуждение 488 и эмиссия 543 нм). Интенсивность флуоресценции определяли количественно, и супероксид-специфическое окрашивание DHE равнялось процентной интенсивности окрашивания DHE минус процентной интенсивности окрашивания DHE в присутствии темпола.
Анализы сосудистой реактивности
Мышей ApoE-KO, получавших WD, лечили в течение 28 дней CavNOxin или пептидами-носителями.После этого грудную аорту вырезали и разрезали на сегменты длиной 4 мм. Кольца подвешивались на двух вольфрамовых проволоках, установленных в системе миографа сосудов (Danish Myotechnologies, Орхус, Дания) в насыщенном кислородом буфере Кребса при 37 ° C, как опубликовано (25). Чтобы изучить NOS-специфичность этих васкоконстрикторных ответов, кольца предварительно ограничивали субмаксимальной концентрацией фенилэфрина (PE), и L-NAME (100 мкмоль) вводили на плато сокращения, вызванного PE.
Данные и статистический анализ
Все данные представлены как среднее ± стандартная ошибка среднего и считаются статистически значимыми при P <0.05 с помощью однофакторного дисперсионного анализа с последующим апостериорным тестом Ньюмана-Кеулса. Для анализа лейкоцитарно-эндотелиального взаимодействия ex vivo был проведен двухфакторный дисперсионный анализ с последующим апостериорным тестом Бонферрони. Для иммуногистохимического окрашивания VCAM-1, DHE, 4-HNE и NT данные нормализованы для контрольной группы ND для мышей ApoE KO с STZ-индуцированным диабетом и группы WD + носитель для мышей ApoE KO, получавших WD.
Результаты
CavNOxin не влияет на метаболические параметры у мышей с STZ-индуцированным диабетом и мышей ApoE KO, получавших WD
Масса тела, уровень глюкозы в крови и метаболические параметры мышей с STZ-индуцированным диабетом и ApoE KO, получавших WD мыши показаны в таблицах 1 и 2 соответственно.Как и ожидалось, мыши с STZ-индуцированным диабетом и мыши ApoE KO, получавшие WD, демонстрировали повышенные уровни глюкозы, триглицеридов и холестерина натощак по сравнению с ND и мышами ApoE KO, получавшими стандартную пищу, соответственно. Метаболические фенотипы этих мышей были аналогичны T1D и T2D (29). Наиболее важно то, что обработка либо CavNOxin (2,5 или 5 мг / кг), либо пептидом-носителем не изменяла параметры глюкозы или липидов ни в одной из моделей. Как и ожидалось, у мышей с STZ-индуцированным диабетом уровень инсулина в плазме снизился на ~ 65% по сравнению с мышами ND ApoE KO, без эффекта у мышей, получавших CavNOxin (таблица 1).Мыши ApoE KO, получавшие WD, демонстрировали тенденцию к повышению артериального давления, тогда как индукция STZ не влияла на уровни базального артериального давления (дополнительный рисунок 4). Обработка CavNOxin не повлияла на артериальное давление в обеих моделях.
Таблица 1Масса тела, уровень глюкозы в крови, гликированный гемоглобин, триглицериды, холестерин, ЛПВП, холестерин не-ЛПВП, соотношение ЛГ и инсулин в плазме (нг / мл) определялись через 10 недель диабета в присутствии или в отсутствие лечения CavNOxin. у STZ-индуцированных мышей ApoE KO
Таблица 2Масса тела, уровень глюкозы в крови, триглицериды, холестерин, ЛПВП, холестерин не-ЛПВП и соотношение ЛГ определяли через 12 недель стандартной еды или WD в присутствии или в отсутствие обработки CavNOxin
CavNOxin защищает от STZ- и WD-индуцированного атеросклероза
STZ-индуцированных мышей ApoE KO показали значимое значение 9.2-кратное увеличение общей площади бляшек по сравнению с мышами ND ApoE KO (рис. 1 A и репрезентативные изображения на дополнительном рис. 1), при этом увеличенное количество бляшек наблюдается в дуге, грудной клетке и брюшной полости (9,2-, В 9,2 и 7,5 раза соответственно) (рис. 1 B – D ). Лечение CavNOxin в дозе 5 мг / кг ослабило общую бляшку аорты на 70% (рис. 1 A – D ) (площадь бляшки на дуге уменьшилась на 80%, площадь грудной бляшки уменьшилась на 84%), хотя уменьшения бляшки на брюшной аорте не произошло. существенный.Более низкая доза CavNOxin (2,5 мг / кг) снизила общую бляшку на 56% (рис. 1 A ), что отражает дозозависимость. У STZ-индуцированных мышей ApoE KO площадь поражения синуса аорты увеличилась в 2,4 раза по сравнению с мышами ND (рис. 1 E ), которая уменьшилась на 81% после обработки CavNOxin (5,0 мг / кг) (рис. 1 E ) (526 704 ± 34 261 против 282 586 ± 23 635 мкм 2 ).
Рисунок 1CavNOxin снижает атеросклероз у мышей ApoE KO, индуцированных STZ и получавших WD. Количественная оценка атеросклеротических поражений в общей аорте, дуге, грудном отделе, брюшной полости и синусе аорты от ND (белый), STZ-индуцированного диабета (серый), диабетика + носитель (черный), диабетика + CavNOxin– (2.5 мг / кг; вертикальная полоса) и мышей ApoE KO с диабетом + CavNOxin (5,0 мг / кг; диагональная полоса) показаны в A — E соответственно. *** P <0,001 по сравнению с группой ND, # P <0,05, ## P <0,01 по сравнению с группой диабетиков и диабетиков + носитель, ### P <0,001 по сравнению с группой диабетиков + носитель . Количественная оценка атеросклеротических поражений в общей аорте, дуге, грудной клетке, брюшной полости и синусе аорты при кормлении кормом + носитель (белый), при кормлении кормом + обработанный CavNOxin (серый), на кормлении WD + обработанный носителем (черный ) и мышей ApoE KO, получавших кормление WD + CavNOxin (горизонтальная полоса), показаны в F — J соответственно.* P <0,05, ** P <0,01, *** P <0,001 по сравнению с кормом + носитель; # P <0,05, ## P <0,01, ### P <0,001 по сравнению с носителем WD +. n = 5–10 на группу (для A – E ) и n = 10 на группу (для F – J ). Все результаты выражены как среднее ± SEM.
После 12 недель WD процент бляшек в общей части аорты, дуги, грудной клетки и брюшной полости, а также в синусе аорты увеличился 6.В 4, 7,4, 8,5, 4,0 и 2,8 раза по сравнению со стандартным кормлением мышей ApoE KO, получавшими носитель (рис. 1 F – J и репрезентативные изображения на дополнительном рис. 2). Лечение CavNOxin не имело эффекта у мышей, получавших стандартную пищу (рис. 1 F – J ). Однако у мышей ApoE KO, получавших WD, обработка CavNOxin приводила к 48% уменьшению общей площади аортальной бляшки по сравнению с получавшими носитель мышами ApoE KO, получавшими WD (фиг.1 F ) (площадь поражения: 6,80 ± 0,48 против 3,98 ± 0,56%). Сечения дуги, грудной и брюшной аорты показали снижение атеросклероза на 50, 48 и 47% соответственно (рис.1 G – I ), тогда как синусы аорты показали уменьшение площади поражения на 35% (Рис. 1 J ) (386 612 ± 24 044 мкм 2 против 299 896 ± 21 607 мкм 2 площади поражения). Эти данные демонстрируют, что CavNOxin обеспечивает высокую степень атеропротекции у мышей ApoE KO, индуцированных STZ и получавших WD.
CavNOxin улучшает функцию эндотелия и снижает маркеры окислительного стресса
Чтобы подтвердить улучшение функции эндотелия в присутствии атерогенных факторов, мышей ApoE-KO, получавших WD, лечили CavNOxin или пептидами-носителями в течение 28 дней (до каких-либо серьезных морфологических изменений сосудов) , а кольца аорты устанавливали на миограф (25).Стимуляция PE (10 −9 −10 −4 моль) (рис. 2 A ) вызвала зависимое от концентрации сужение аорт, обработанных носителем, тогда как сосуды, обработанные CavNOxin, показали более низкое среднее сужение, что свидетельствует об улучшении функции эндотелия. . Мыши, получавшие CavNOxin, демонстрировали повышенную регуляцию функции NO, потому что они демонстрировали большее L-NAME-индуцированное увеличение сокращения PE (Fig. 2 B ). Кроме того, были измерены нитриты / нитраты плазмы, стабильные продукты окисления NO.Мыши ApoE KO, получавшие WD, показали уровни нитритов / нитратов в плазме, аналогичные таковым у их коллег, получавших стандартную пищу (рис. 2 D ). 12-недельная обработка CavNOxin привела к значительному увеличению уровней нитритов в плазме у мышей ApoE KO, получавших WD, по сравнению с мышами ApoE KO, получавшими WD, получавших WD (фиг. 2 D ) ( P <0,05), указывает на положительную регуляцию биодоступности NO. Индукция STZ привела к незначительному снижению уровней нитритов в плазме, тогда как у мышей ApoE KO, обработанных CavNOxin, STZ-индуцированные уровни нитритов вернулись к уровням их коллег ND (рис.2 C ) ( P = несущественно).
Рисунок 2CavNOxin увеличивает функцию эндотелия и снижает окислительный стресс in vivo. A и B : аорты, обработанные CavNOxin, от мышей ApoE KO, получавших WD, демонстрируют уменьшенные PE-индуцированные сужения (мН) (квадраты) по сравнению с аортами, обработанными носителем (треугольник). B : Среднее увеличение сужения, вызванное L-NAME (10 -6 моль). n = 4 / группа. * P <0,05.Общая концентрация нитрита в плазме показана для мышей ApoE KO с STZ-индуцированным диабетом ( C ) и для мышей ApoE KO, получавших WD ( D ), получавших носитель или CavNOxin. * P <0,05 по сравнению с кормом + носитель. Анализ dROMs показан у мышей ApoE KO с STZ-индуцированным диабетом ( E ) и мышей ApoE KO, получавших WD ( F ), получавших носитель или CavNOxin. Для C и E столбцы представляют собой ND (белый), STZ-индуцированный диабет (серый), диабет + лечение носителем (черный), диабет + CavNOxin (2.5 мг / кг) (вертикальная полоса) и диабетических + CavNOxin (5,0 мг / кг) (диагональная полоса) ApoE KO мышей. *** P <0,01 по сравнению или ND для корма ( E ) + носитель для F , # P <0,05 по сравнению с диабетом и диабетиком + носитель. n = 7–10 на группу (для C и E ) и n = 10 на группу (для D и F ). Все результаты выражены как среднее ± SEM.
Поскольку улучшение функции эндотелия обычно связано со снижением окислительного стресса и АФК, таких как супероксид и пероксинитрит (35), мы проанализировали свободные радикалы / гидропероксиды в плазме, а также уровни нитротирозина в аорте, DHE и 4-HNE, которые являются маркеры опосредованного пероксинитритом повреждения, супероксид и побочный продукт перекисного окисления липидов, соответственно, при иммуногистохимическом окрашивании.Во-первых, свободные радикалы / гидропероксиды (тест dROMs) были увеличены у STZ-индуцированных мышей ApoE KO и мышей ApoE KO, получавших WD, по сравнению с ND и мышами, получавшими стандартную пищу, соответственно (рис. 2 E и F ). (Единицы Каррателли). CavNOxin снижал уровни dROM у мышей ApoE KO с STZ-индуцированным диабетом при обеих дозах (фиг. 2 E ) ( P <0,05). Напротив, CavNOxin не влиял на уровни dROM у мышей, получавших WD (фиг. 2 F ). Мы наблюдали, что уровни нитротирозина были значительно снижены до 56% у STZ-индуцированных мышей ApoE KO (рис.3 A ) и 52% у мышей ApoE KO, получавших WD (фиг. 3 B ), получавших CavNOxin, по сравнению с мышами, получавшими носитель. Точно так же супероксид-специфическое окрашивание DHE было значительно снижено на ~ 33% у STZ-индуцированных мышей ApoE KO и на 66% у мышей ApoE KO, получавших WD, получавших CavNOxin, по сравнению с мышами, получавшими носитель (фиг. 3 C). и D ). Наконец, CavNOxin также уменьшал окрашивание 4-HNE на 69% у мышей ApoE KO, индуцированных STZ, без значительных различий у мышей ApoE KO, получавших WD (фиг.3 E и F ). Более того, уровни 8-изопростана в моче, маркер окислительного стресса, были нормализованы обратно к уровням ND у STZ-индуцированных мышей ApoE KO, получавших CavNOxin (дополнительный рисунок 3 C ), тогда как WD не оказал никакого эффекта (данные не показаны) . Наконец, базальная продукция супероксида в эндотелиальных клетках, обработанных носителем или CavNOxin (1 и 5 мкмоль), выявила значительно сниженные базальные уровни супероксида в клетках, обработанных CavNOxin, по сравнению с клетками, обработанными носителем (дополнительный рис.3 D ). Взятые вместе, эти данные предполагают, что CavNOxin обеспечивает атерозащиту за счет снижения окислительного стресса.
Рисунок 3CavNOxin снижает параметры окислительного стресса. Репрезентативные изображения и количественная оценка аорт, окрашенных антителом против NT, от мышей ApoE KO с STZ-индуцированным диабетом ( A ) и мышей ApoE KO, получавших WD ( B ), получавших носитель или CavNOxin. *** P <0,001 по сравнению с группой ND, # P <0,05 по сравнению с группой диабетиков + носитель, ### P <0.001 по сравнению с диабетической группой для A ; ** P <0,01 по сравнению с группой WD + носитель для B . Репрезентативные изображения и количественная оценка обнаружения супероксида с помощью флуоресцентной визуализации DHE и DHE плюс tempol (вставка) в аортах STZ-индуцированных диабетических ( C ) и WD мышей ApoE KO ( D ), получавших носитель или CavNOxin. * P <0,05 по сравнению с ND для C . ** P <0,01 по сравнению с автомобилем WD + для D . Репрезентативные изображения и количественная оценка аорт, окрашенных 4-HNE, у мышей ApoE KO с индуцированным STZ диабетом ( E ) и мышей ApoE KO, получавших питание WD ( F ), получавших носитель или CavNOxin.** P <0,01 по сравнению с группой ND, # P <0,05 по сравнению с группой диабетиков и диабетиков + носитель. Для A , C и E столбцы представляют ND (белый), STZ-индуцированный диабет (серый), диабет + лечение носителем (черный), диабет + CavNOxin (2,5 мг / кг) (вертикальная полоса) и диабетические + CavNOxin (5,0 мг / кг) (диагональная полоса) мышей ApoE KO. n = 5–10 / группа. Все результаты выражены как среднее ± SEM.
CavNOxin снижает проатерогенный эффект VCAM-1
Ослабление функции эндотелия сопровождается активацией эндотелия и повышением активности молекул адгезии.Поэтому мы сравнили экспрессию генов специфических провоспалительных маркеров, перечисленных в дополнительной таблице 1, после лечения CavNOxin. По данным нашего анализа мРНК, VCAM-1, хорошо известный проатерогенный и провоспалительный медиатор в условиях ослабленного образования эндотелиального NO, сильно повлиял в условиях диабета и при лечении CavNOxin. У мышей ApoE KO, индуцированных STZ, почти трехкратное увеличение экспрессии мРНК VCAM-1, вызванное диабетом, было почти полностью нормализовано CavNOxin (рис.4 A ) и снижается на ~ 67% на уровне белка (рис. 4 B ). Обработка CavNOxin приводила к значительному снижению экспрессии гена VCAM-1 в аортах мышей ApoE KO, получавших WD (фиг. 4 C ) (носитель: 1,06 ± 0,17 условных единиц [у.е.] по сравнению с CavNOxin: 0,59 ± 0,05 ед. ) и снижение экспрессии белка VCAM-1 на 64% (фиг. 4 D ). В изолированных эндотелиальных клетках экспрессия белка VCAM-1, индуцированная TNF-α, провоспалительным цитокином, важным при диабете, была снижена на 71% при совпадении пептида CavNOxin (рис.4 E ). Эти данные убедительно свидетельствуют о том, что лечение CavNOxin способствует устойчивому противовоспалительному ответу, возможно, как следствие пониженного окислительного состояния, вызванного CavNOxin. Этот противовоспалительный эффект пептида дополнительно подтверждается значительным снижением окрашивания для маркера макрофагов, F4 / 80, а также снижением экспрессии гена MCP-1 в STZ-индуцированных аортах ApoE KO (дополнительный рисунок 3 A ). и B ).
Фиг.4.CavNOxin снижает экспрессию VCAM-1 у мышей ApoE KO с STZ-индуцированным диабетом и мышей ApoE KO, получавших WD. A : уровни экспрессии мРНК VCAM-1, определенные с помощью qRT-PCR и выраженные как кратность индукции по сравнению с контролями ND. Столбики представляют ND (белый), STZ-индуцированный диабет (серый), диабетик + лечение носителем (черный), диабетик + CavNOxin (вертикальная полоса) и диабетик + CavNOxin (5,0 мг / кг) (диагональная полоса) ApoE KO мышей. B : Типичные изображения и количественная оценка иммуноокрашивания VCAM-1 в аортах мышей ApoE KO с индуцированным STZ диабетом, получавших носитель или CavNOxin (комбинированное значение для обоих 2.5 и 5,0 мг / кг). * P <0,05, *** P <0,001 по сравнению с группой ND, # P <0,05 по сравнению с диабетиком. C : уровни экспрессии мРНК VCAM-1, определенные с помощью qRT-PCR и выраженные как кратность индукции относительно контроля носителя у мышей ApoE KO, получавших WD. D : Типичные изображения и количественная оценка иммуноокрашивания VCAM-1 в аортах мышей ApoE KO, получавших WD, получавших носитель или CavNOxin. * P <0,05, ** P <0,01 по сравнению с группой носителя. E : Вестерн-блоттинг и денситометрия экспрессии белка VCAM-1 относительно β-актина в HAEC, обработанных либо носителем, либо пептидом CavNOxin (5 мкмоль в течение 6 часов) в присутствии TNF-α (0,5 нг / мл). * P <0,05 по сравнению с группой, обработанной TNF-α, ** P <0,01 по сравнению с группой, обработанной носителем + TNF-α. n = 5–10 / группа. Столбики представляют собой контрольные (белые), обработанные TNF-α (серые), обработанные TNF-α + носителем (черные) и обработанные TNF-α + CavNOxin (горизонтальная полоса) эндотелиальные клетки.Все результаты выражены как среднее ± SEM. Ctrl, контроль.
CavNOxin снижает моноцитарно-эндотелиальные взаимодействия In vitro
Поскольку ослабление уровней VCAM-1 при лечении CavNOxin должно приводить к снижению взаимодействий лейкоцит-эндотелиальные клетки, культивированные HAEC предварительно обрабатывали в течение 6 часов CavNOxin (5 мкмоль). % снижение адгезии человеческих моноцитарных клеток THP-1 в статических анализах адгезии (фиг. 5 A ). В условиях высокого уровня глюкозы (30 ммоль) в течение 3 дней наблюдалось увеличение адгезии клеток THP-1 (рис.5 B ) по сравнению с лечением с низким содержанием глюкозы (5 ммоль), тогда как обработка CavNOxin полностью блокировала адгезию клеток THP-1 в результате высокого содержания глюкозы (фиг. 5 B ).
Рисунок 5CavNOxin снижает взаимодействия моноцитов и эндотелиальных клеток in vitro и ex vivo. A : Количественная оценка прикрепленных человеческих моноцитарных клеток THP-1 к HAEC, обработанным носителем или пептидом CavNOxin. * P <0,05 по сравнению с группой носителя. B : Количественное определение адгезивных клеток THP-1 к HAEC, выращенных в среде с низким содержанием глюкозы (5 ммоль) или высоким содержанием глюкозы (30 ммоль) и обработанных носителем или пептидом CavNOxin.* P <0,05, как указано. Полоски обозначают низкий уровень глюкозы (LG) (белый), LG + обработанный носителем (серый), LG + обработанный CavNOxin (черный), высокий уровень глюкозы (HG) (вертикальная полоса), HG + обработанный носителем (серая вертикальная полоса), и эндотелиальные клетки, обработанные HG + CavNOxin (серый узор). C : Типичные изображения, показывающие связывание лейкоцитов (стрелки) с поверхностью аорты через 15 минут после стимуляции TNF-α. D : Количественное определение среднего связывания лейкоцитов на поле каждые 5 минут в течение 15-минутного периода.** P <0,05, *** P <0,001 по сравнению с группой носителя, # P <0,05, ## P <0,01 по сравнению с группой, обработанной носителем + TNF-α. E : Типичные изображения, показывающие лейкоциты (стрелки), связывающиеся с поверхностью аорты диабетической аорты и аорты ND в 15-минутный момент времени. F : Количественное определение среднего связывания лейкоцитов на поле каждые 5 минут в течение 15-минутного периода. ** P <0,01 по сравнению с группой ND + носитель. # P <0,05, ### P <0.01 по сравнению с группой, получавшей диабет + CavNOxin. n = 6–8 / группа. Все результаты выражены как среднее ± SEM.
CavNOxin снижает динамическую адгезию сосудов аорты Ex vivo
Чтобы подтвердить in vitro эффекты CavNOxin на адгезию лейкоцитов и эндотелия, мы исследовали адгезию лейкоцитов человека к изолированным аортам. В базовых условиях не наблюдалось различий в количестве прикрепленных лейкоцитов между аортами ApoE KO, обработанными носителем или пептидами CavNOxin (рис.5 C и D ). TNF-α (5 нг / мл; 4 ч) индуцировал значительное зависящее от времени увеличение количества прикрепившихся лейкоцитов через 10 и 15 минут (рис. 5 C и D ), которое почти полностью исчезло в аорте. совпадали с CavNOxin (фиг. 5 C и D ) (наполнитель + TNF: 16 ± 3 клеток по сравнению с CavNOxin + TNF: 8 ± 2 клетки через 15 мин). Точно так же диабетическая аорта показала более высокое количество прикрепленных лейкоцитов (рис. 5 E и F ) (диабетический vs.ND), тогда как, что важно, индуцированная диабетом адгезия полностью блокировалась обработкой CavNOxin (рис. 5 E и F ) (диабетик: 15 ± 2 клеток против 5 ± 2 клеток CavNOxin через 15 мин). Наши данные in vitro и ex vivo демонстрируют, что CavNOxin блокирует лейкоцитарно-эндотелиальные взаимодействия, ключевой этап в раннем атерогенезе.
Мыши ApoE KO, лишенные гена eNOS, устойчивы к лечению CavNOxin
Чтобы подтвердить, что CavNOxin опосредует свой терапевтический эффект через eNOS, мы создали мышей ApoE / eNOS dKO.При кормлении WD эти мыши показали трехкратное увеличение атеросклеротической нагрузки по сравнению с мышами ApoE KO, получавшими тот же WD (ApoE KO: 6,8% общего бляшки [см. Фиг. 1 A ] по сравнению с ApoE / eNOS dKO: 18% общего бляшки. [Рис. 6 B ]). Интересно, что лечение носителем или пептидом CavNOxin (фиг. 6 A – E ) приводило к аналогичным атеросклеротическим повреждениям в WD ApoE / eNOS dKO и аналогичному окрашиванию нитротирозином (фиг. 6 F ). Это отсутствие эффекта пептида CavNOxin на атеросклеротические поражения у мышей ApoE / eNOS dKO является постоянным с высокой степенью специфичности пептида CavNOxin в отношении eNOS и указывает на то, что CavNOxin, вероятно, снижает вызванный eNOS окислительный стресс в дополнение к увеличению вызванного eNOS НЕТ, что приводит к атерозащите.
Рисунок 6CavNOxin не оказывает антиатеросклеротического действия у мышей eNOS / ApoE dKO. A : Судан IV-окрашенные аорты от мышей eNOS / ApoE dKO, получавших WD, получавших носитель или пептиды CavNOxin. Процент общей площади бляшки в аорте, процент площади бляшки дуги, процент площади грудной бляшки и процент площади брюшной бляшки показаны в B — E соответственно. F : Количественное определение аорт, окрашенных антителом против NT, у мышей ApoE KO, получавших WD, и мышей eNOS / ApoE dKO, получавших WD, получавших носитель или CavNOxin. n = от 5 до 6 на группу. Все результаты выражены как среднее ± SEM.
Обсуждение
Наше исследование впервые демонстрирует in vivo на двух экспериментальных моделях диабета, а именно на модели STZ-индуцированного T1D и T2D-индуцированной WD инсулинорезистентной модели, которые освобождают eNOS от ингибирующего зажима Cav. -1 приводит к заметному ослаблению атеросклероза. Общепринято, что эндотелиальная дисфункция с сопутствующей ей пониженной биодоступностью NO вносит вклад в ранний атерогенез при диабете, главным образом за счет модуляции окислительного стресса и лейкоцитарно-эндотелиальных взаимодействий.Используя CavNOxin, проницаемый для клеток пептид Cav-1 с инактивированным ингибиторным доменом eNOS, мы демонстрируем, что увеличение высвобождения NO, происходящего из эндогенного eNOS, ослабляет индуцированные диабетом атеросклеротические бляшки, что коррелирует со снижением маркеров окислительного стресса, уменьшением провоспалительные медиаторы и уменьшение прилипания лейкоцитов к эндотелию сосудов. Кроме того, CavNOxin не влиял на атеросклероз у мышей ApoE / eNOS dKO, что подтверждает специфичность нашего подхода к активации eNOS — ключевой результат, поскольку NO также продуцируется NOS нейронов и индуцибельным NO, а экспрессия обеих изоформ была показано, что его активность повышается в атеросклеротических бляшках на поздних стадиях (18).
Биодоступность NO критически зависит от тонкого баланса между синтезом NO на основе eNOS и супероксидной инактивацией NO, причем баланс склоняется в пользу последнего в условиях диабета. Следовательно, ограничение продукции ROS является важным терапевтическим средством повышения биодоступности NO. В нашем исследовании системный окислительный стресс и повреждение тканей, связанное с АФК, было обнаружено в обеих моделях, что коррелировало с увеличением атеросклеротической нагрузки. Это наблюдение подтверждается исследованиями на людях, которые показывают высокие уровни конкретного биомаркера липидпероксидазы, отражающего окислительный стресс в популяции диабетиков (36).Недавние исследования показали, что основным источником АФК при диабетических макрососудистых заболеваниях является несвязанная eNOS. Действительно, разобщение eNOS настолько глубоко в диабетическом эндотелии, что полученный из eNOS супероксид вызывает усиление ремоделирования и дисфункции сосудов, укрепляя представление о том, что правильная регуляция eNOS необходима для защиты от атеросклероза, особенно в диабетической среде (37). В этом исследовании мы продемонстрировали новый антиоксидантный эффект CavNOxin, поскольку он способен значительно снизить системный окислительный стресс, а также повреждение сосудистой ткани, вызванное пероксинитритом и перекисным окислением липидов.Мы предполагаем, что снижение окислительного стресса, наблюдаемое в этом исследовании, является результатом действия CavNOxin на передачу сигналов eNOS, что отражается в неспособности CavNOxin оказывать антиатеросклеротические эффекты у мышей ApoE / eNOS dKO.
Центральным механизмом, вызывающим сосудистые заболевания, связанные с диабетом, является воспаление, которое запускает эндотелиальную дисфункцию и усиливает проадгезивные и протромботические свойства эндотелия, тем самым способствуя привлечению воспалительных клеток к сосудистой стенке.Другой ключевой вывод нашей работы связан с наблюдением, что индуцированная CavNOxin повышающая регуляция NO способна ослаблять экспрессию VCAM-1 в аорте и эндотелия, что тесно коррелирует со снижением адгезии лейкоцитов / моноцитов к сосудистой стенке и культивированным эндотелиальным клеткам у больных диабетом. и воспалительные (TNF-α) параметры, подтверждая предыдущие исследования, которые продемонстрировали снижение рекрутирования лейкоцитов и экспрессии VCAM-1 в тканях KO Cav-1 (38-40). Более того, повышенная активность eNOS была связана с подавлением активности молекул адгезии, в частности VCAM-1 (19), и наши данные предполагают, что это может быть достигнуто специфически за счет эндогенного eNOS компенсаторным образом, поскольку гипергликемия является мощным индуктором лейкоцитов. эндотелиальные взаимодействия (13,41).Кроме того, известно, что окислительный стресс регулирует провоспалительные медиаторы, такие как VCAM-1 (42). Учитывая положительные эффекты CavNOxin в снижении уровня супероксида, вполне возможно, что снижение уровней VCAM-1 происходит в ответ на пониженный окислительный стресс, вызванный CavNOxin. Также возможно, что CavNOxin опосредует свое противовоспалительное действие на VCAM-1 посредством модификаций кровотока, которые, как известно, влияют на уровни VCAM-1 (43).
Хотя снижение биодоступности NO, полученного из eNOS, долгое время связывали с увеличением частоты и тяжести сердечно-сосудистых заболеваний, неизвестно, можно ли стимулировать высвобождение эндогенного NO, происходящего из eNOS, специально для усиления атеропротекции.Как отмечалось ранее, потенциальное действие по повышению биодоступности eNOS действительно может быть особенно актуальным при диабете, который является состоянием дефицита NO. Несколько исследований подтвердили возможность увеличения высвобождения NO, происходящего из eNOS, за счет уменьшения взаимодействия eNOS с Cav-1 (44–47). Однако в этих предыдущих исследованиях не использовались фармакологические агенты, специфичные для NOS, такие как ингибиторы HMGCoA редуктазы (статины) или блокатор каналов Ca 2+ гладкомышечных клеток (амлодипин) в неатерогенных или диабетических условиях.Сердечно-сосудистые препараты, такие как статины, ингибиторы АПФ, блокаторы рецепторов ангиотензина, β-адреноблокаторы (обзор в 48) или добавление BH 4 (49), могут увеличивать высвобождение NO в клинических или доклинических условиях, но это скорее плейотропные или вторичные эффекты. чем прямая модуляция NO, происходящего из eNOS. Высокая степень специфичности CavNOxin в отношении eNOS позволяет нам утверждать, что в пораженных сосудах существует атеропротекторная, производная eNOS эндотелиальная резервная функция, несмотря на присутствие атерогенных факторов, вызывающих эндотелиальную дисфункцию.Таким образом, наши данные показали, что прямое улучшение функции эндотелия за счет специфического воздействия на eNOS через механизм, зависимый от eNOS / Cav-1, приводит к атерозащите при атеросклерозе, индуцированном диабетом. Что еще более важно, уникальная способность CavNOxin регулировать активность eNOS при сохранении биологии кавеол / Cav-1 делает его желательным терапевтическим агентом. Однако в настоящее время требуется дальнейшая валидация на моделях T1D и T2D, особенно для тех, у кого дислипидемия не может играть столь заметную роль, например, индуцированный STZ LDL-R — / — мыши (для T1D) или db / db модель T2D.Кроме того, наши данные предполагают, что потенциальное нацеливание на этот сайт с помощью новых агонистических пептидов или малых молекул является многообещающим в качестве таргетной антиатерогенной терапии в условиях высокого уровня глюкозы и повышенных липидов, как это наблюдается у пациентов с диабетом.
Информация об изделии
Благодарности. Авторы благодарят лабораторию сосудистой фармакологии профессора Джей Чин-Дастинг из Института сердца и диабета Baker IDI (Мельбурн) за разрешение использовать оборудование для исследований адгезии потока и Ирен Кармайкл из Monash Micro Imaging из Центра медицинских исследований и образования Альфреда. (Мельбурн) за помощь в визуализации DHE.
Финансирование. J.B.d.H. был поддержан грантом 1005851 Австралийского национального совета по здравоохранению и медицинским исследованиям. поддерживается грантами Канадских институтов исследований в области здравоохранения, Фонда сердца и инсульта Канады, Фонда сердца и инсульта Британской Колумбии и Юкона, Фонда Майкла Смита исследований в области здравоохранения, Канадского фонда инноваций и Фонда развития знаний Британской Колумбии. В КАЧЕСТВЕ. поддерживается стипендией Александра Грэхема Белла для аспирантов в Канаде, четырехлетней докторской стипендией Канадских институтов исследований в области здравоохранения / Университета Британской Колумбии и дополнительной стипендией Baker IDI.Эта работа была частично поддержана Программой поддержки оперативной инфраструктуры правительства штата Виктория.
Двойственность интересов. О потенциальных конфликтах интересов, относящихся к этой статье, не сообщалось.
Авторские взносы. A.S. исследовал данные, разработал дизайн исследования и написал рукопись. S.S., N.S., C.L., S.M.T. и O.H. исследованные данные. D.J.G. просмотрел и отредактировал рукопись. M.E.C. участвовал в обсуждении и рецензировал отредактированную рукопись.J.B.d.H. и П. разработал дизайн исследования и проверил отредактированную рукопись. В КАЧЕСТВЕ. является гарантом этой работы и, как таковой, имеет полный доступ ко всем данным в исследовании и берет на себя ответственность за целостность данных и точность анализа данных.
- Получено 7 апреля 2015 г.
- Принято 20 июня 2015 г.
- © 2015 Американской диабетической ассоциацией. Читатели могут использовать эту статью при условии, что произведение правильно процитировано, используется в образовательных целях, а не для получения прибыли, и если работа не изменена.
Дольчатое связывание кальция в кальмодулине регулирует активацию эндотелиальной синтазы оксида азота
Абстрактные
Фон
Эндотелиальной синтазе оксида азота (eNOS) человека для переноса электронов требуется кальций-связанный кальмодулин (CaM), но подробный механизм остается неясным.
Методология / основные выводы
Используя серию мутантов CaM с заменой E на Q в четырех сайтах связывания кальция, мы обнаружили, что единичная мутация в любом сайте связывания кальция (B1Q, B2Q, B3Q и B4Q) приводила к ~ 2-3-кратному увеличению Концентрация CaM, необходимая для полумаксимальной активации (EC50) образования цитруллина, что указывает на то, что каждый кальций-связывающий сайт CaM вносит вклад в ассоциацию между CaM и eNOS.Анализы образования цитруллина и восстановления цитохрома с показали, что по сравнению с nNOS или iNOS, eNOS менее требовательна к связыванию кальция с каждым из четырех сайтов связывания кальция. Однако специфическое для долей разрушение с двойными мутациями в сайтах связывания кальция либо в N- (B12Q), либо в C-концевых (B34Q) долях значительно снижает как оксигеназную, так и редуктазную активности eNOS. Анализ сдвига подвижности геля и измерение флуоресценции флавина показали, что N- и C-доли СаМ играют разные роли в регулировании катализа eNOS; С-концевые EF-руки в его связанной с кальцием форме были ответственны за связывание канонического СаМ-связывающего домена, тогда как N-концевые EF-руки в его связанной с кальцием форме контролировали перемещение домена FMN.Ограниченные исследования протеолиза также продемонстрировали, что B12Q и B34Q вызывают различные конформационные изменения в eNOS.
Выводы
Наши результаты ясно демонстрируют, что CaM контролирует перенос электронов eNOS, прежде всего, посредством его специфичного для долей связывания кальция.
Образец цитирования: Wu P-R, Kuo C-C, Yet S-F, Liou J-Y, Wu KK, Chen P-F (2012) Долевое связывание кальция в кальмодулине регулирует активацию синтазы оксида азота эндотелия. PLoS ONE 7 (6): e39851.https://doi.org/10.1371/journal.pone.0039851
Редактор: Андреас Хофманн, Университет Гриффита, Австралия
Поступила: 16 марта 2012 г .; Одобрена: 31 мая 2012 г .; Опубликован: 29 июня 2012 г.
Авторские права: © 2012 Wu et al. Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Финансирование: Работа была поддержана грантом национальных научно-исследовательских институтов здравоохранения (99A1-CSAP01-014) на Тайване. Финансирующие организации не играли никакой роли в дизайне исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи.
Конкурирующие интересы: Авторы заявили об отсутствии конкурирующих интересов.
Введение
Эндотелиальная синтаза оксида азота человека (eNOS) катализирует синтез оксида азота (• NO), который является ключевым регулятором сердечно-сосудистого гомеостаза [1], [2].Подобно нейронной NOS (nNOS) [3], [4] и индуцируемой макрофагами NOS (iNOS) [5], [6], eNOS [7] является гомодимером; каждый мономер содержит С-концевой домен редуктазы и N-концевой домен оксигеназы [8] — [10]. Домен редуктазы связывает кофакторы FAD и FMN и состоит из сайта связывания NADPH, в то время как домен оксигеназы связывает гем протопорфрина IX, тетрагидробиоптерин (H 4 B) и содержит сайт связывания L-аргинина. Эти два домена связаны канонической последовательностью связывания CaM.Связывание кальция / СаМ с этим сайтом способствует внутридоменному переносу электронов между FAD и FMN, а также междоменному переносу электронов от флавина одной субъединицы к гему другой субъединицы [11] — [13]. Хотя механизм, с помощью которого CaM регулирует активацию NOS, интенсивно исследуется, многое остается нерешенным. Наша недавняя работа над eNOS продемонстрировала, что в дополнение к предполагаемому связывающему домену CaM (остатки 491–510, далее обозначаемые просто как CBD) [14], есть два других участка, i.е. остатка 174–193 сайта связывания гема и остатки 729–757, расположенные в шарнирной области, соединяющей субдомены FAD и FMN, возможно, участвуют в регулируемой CaM / кальцием активации и дезактивации eNOS [15]. Таким образом, механизмы, лежащие в основе CaM / кальций-зависимого катализа eNOS, более сложны, чем считалось ранее.
CaM — это повсеместный кальций-связывающий белок, структурно напоминающий гантель. Он содержит N- и C-концевые доли, соединенные гибким спиральным линкером [16].Каждая доля состоит из двух рук E – F с двумя сайтами связывания кальция. Сайты связывания кальция в C-концевой доле (сайты 3 и 4) имеют более высокое сродство к кальцию, чем сайты в N-концевой доле (сайты 1 и 2) [17]. Связывание кальция запускает конформационные изменения CaM, сводя две доли вместе, образуя гидрофобный интерфейс для взаимодействия с его белками-мишенями [18].
CaM регулирует все три изоформы NOS. iNOS прочно связывает CaM при низком уровне кальция и является каталитически активным при базальных концентрациях кальция в клетке [19], в то время как eNOS и nNOS связывают CaM обратимо в зависимости от концентрации кальция и требуют высоких концентраций кальция для катализа [7], [20].Предыдущие сообщения об использовании мутантов CaM с разными мутациями сайтов связывания кальция предполагают различную роль четырех сайтов связывания кальция для nNOS и iNOS в регуляции связывания CaM и переноса электронов [21] — [23]. Роль, которую каждый кальций-связывающий сайт играет в регуляции связывания CaM с eNOS и каталитической активностью eNOS, неясна. Поскольку eNOS прикрепляется к плазматической мембране посредством миристоилирования и пальмитайлирования, кальций-CaM-зависимый перенос электронов и катализ, вероятно, будут зависеть от различных сигнальных путей, что делает его более сложным, чем iNOS или nNOS [24].Кроме того, поскольку кальций-связывающие сайты СаМ могут по-разному регулировать функцию целевого белка, а большая часть свободного СаМ не полностью занята четырьмя ионами кальция в колеблющейся внутриклеточной концентрации кальция [25], важно уточнить роль каждого из них. кальций-связывающий сайт CaM в активности eNOS.
Рисунок 1. Мутантные конструкции CaM.
A: Показана последовательность человеческого CaM с использованием однобуквенных кодов аминокислотных остатков. Мутировавший аминокислотный остаток в каждом сайте связывания кальция отмечен красным.Спиральные области показаны курсивом и синим цветом. B: Электрофоретическая подвижность белков CaM дикого типа и мутантных белков была проанализирована с помощью SDS-PAGE. Образец 5 мкг каждой конструкции CaM загружали в 15% SDS-PAGE, который проводили со стандартным буфером Laemmli SDS-PAGE в присутствии 1 мМ EGTA. Молекулярные массы стандартов белка показаны слева в килодальтонах (кДа).
https://doi.org/10.1371/journal.pone.0039851.g001
Электронный поток в изоформах NOS контролируется связыванием CaM / кальция.В отсутствие CaM домен FMN находится в экранированном состоянии (вход), который заблокирован связыванием NADPH [26] и стабилизирован относительным пространственным расположением аутоингибиторных элементов и С-концевого хвоста. В этом состоянии домен FMN находится в тесном контакте со связывающим доменом FAD / NADPH [27], и перенос электронов ограничивается FAD к FMN, а не к другим акцепторам электронов. Связывание CaM / кальция вызывает поворот FMN-домена в сторону деэкранированного (выходного) состояния, что облегчает перенос электронов на гем / H 4 B или цитохром c [28] — [31].N- и C-концевые доли CaM связывают ионы кальция с различным сродством, и функции CaM, специфичные для долей, часто наблюдаются в регуляции его белков-мишеней [32], [33]. Мы предположили, что смещение субдомена FMN от входных к выходным состояниям для переноса электронов зависит от специфичного для долей связывания кальция / CaM с eNOS. Чтобы проверить эту гипотезу, мы мутировали один сайт связывания кальция (обозначенный B 1Q , B 2Q , B 3Q и B 4Q соответственно), оба сайта связывания в N-доле (B 12Q ) или C-доли (B 34Q ) или всех четырех сайтов связывания кальция (B 1234Q ), и оценили, как эти мутации влияют на связывание CaM с доменом связывания CaM и опосредованное CaM изменение конформации eNOS и каталитическую активность.
Результаты
мутантов CaM в активации eNOS
мутантов CaM, несущих различные замены E на Q в сайтах связывания кальция, были созданы на основе последовательности CaM человека, показанной на рисунке 1A. Праймеры, использованные для создания мутантов CaM, и обозначенные названия для каждого мутанта были перечислены в таблице 1. Очищенные мутанты CaM имели чистоту не менее 95% с расчетной молекулярной массой ~ 17 кДа на основе анализа SDS-PAGE, который выполняли в стандарте. Буфер SDS-PAGE, содержащий 1 мМ EGTA.Обычно мы получали от 16 до 100 мг очищенных белков из литра питательной среды. Электрофоретическая подвижность большинства мутантов явно не отличалась от таковой у СаМ дикого типа, за исключением мутанта B 4Q , который имел немного более высокую подвижность (рис. 1B).
Стационарное образование цитруллина использовали для определения потока электронов от НАДФН к гему оксигеназного домена. Эффекты СаМ дикого типа и мутантных СаМ на образование цитруллина оценивали путем добавления возрастающих концентраций СаМ к смеси для анализа.Кривые зависимости реакции от концентрации для СаМ дикого типа и каждого мутанта показаны на рисунке 2. По сравнению с СаМ дикого типа, B 1234Q не показал никакой активации, в то время как B 12Q и B 34Q значительно уменьшили способность активировать образование цитруллина. через их широкую концентрацию. Кривые ответа четырех односайтовых мутантов (B 1Q , B 2Q , B 3Q и B 4Q ) напоминали кривую ответа дикого типа. Количественный анализ нескольких экспериментов показал, что ЕС 50 (концентрация, необходимая для половинного максимального образования цитруллина) для односайтовых мутантов примерно в 2–3 раза выше, чем у мутантов дикого типа (Таблица 2).Эти данные предполагают, что каждый из четырех сайтов связывания кальция поддерживает ассоциацию между CaM и eNOS (Таблица 2).
Рис. 2. Образование цитруллина в зависимости от концентрации CaM.
Цитруллин-образующую активность определяли в реакционной смеси, содержащей 25 мМ Трис, pH 7,5, 100 нМ eNOS, 3H-L-аргинин ( 1 мкКи), L-аргинин (20 мкМ), НАДФН (100 мкМ M ), H 4 B (10 мкМ, M) и CaCl 2 (300 мкМ), 0,2 мМ ЭДТА, 100 мМ NaCl, 0.1 мМ DTT, 10% глицерин и указанная концентрация каждой конструкции CaM, как описано в экспериментальных процедурах. Данные представляют собой среднее значение повторяющихся измерений с отклонением <5%. Каждый эксперимент повторяли трижды.
https://doi.org/10.1371/journal.pone.0039851.g002
По мере увеличения концентрации СаМ некоторые мутанты проявляли цитруллин-образующую активность даже выше, чем СаМ дикого типа. Мы сравнили образование цитруллина в присутствии насыщающих концентраций для каждой конструкции CaM (0.5 мкМ), и активность, полученная с eNOS, связанной с СаМ дикого типа, была установлена на 100%. Как показано на Фигуре 3А, СаМ дикого типа увеличивал образование цитруллина в ~ 50 раз по сравнению с тем, что наблюдалось в отсутствие СаМ. Максимальное образование цитруллина с B 1Q было снижено на 25%, а B 2Q существенно не отличалось от дикого типа (Рисунок 3A), в то время как максимальное образование цитруллина для B 3Q и B 4Q было немного выше, чем что дикого типа. Из двух двойных мутантов степень максимальной активности с B 12Q и B 34Q была снижена до 24% и 18% от дикого типа, соответственно.Активность с B 1234Q не увеличилась и осталась на уровне без CaM (рис. 3A). Результат показывает, что, за исключением B 1Q , одиночная мутация в кальций-связывающем сайте CaM мало влияет на активность образования цитруллина eNOS, в то время как нарушение в двух кальций-связывающих сайтах либо в N- (B 12Q ), либо в C- Терминальная доля (B 34Q ) сильно влияет на активность оксигеназы eNOS.
Рисунок 3. Способность различных мутантов CaM активировать ферменты eNOS.
A: Активность образования цитруллина. B: Цитохром c восстановительная активность. И образование цитруллина, и восстановление цитохрома с измеряли в присутствии насыщающей концентрации каждой конструкции СаМ (0,5 мкМ). Активности NOS выражали как процент от соответствующей максимальной активности NOS (100%), которая была получена с eNOS, связанным с CaM дикого типа. В этих условиях активность eNOS, связанного с СаМ дикого типа, составляла ~ 14 мин -1 для образования цитруллина и ~ 286 мин -1 для восстановления цитохрома с .Данные представлены как среднее ± стандартное отклонение. * обозначает p < 0,05, ** <0,01, NS, несущественная разница по сравнению с CaM дикого типа. Каждый эксперимент проводили в трех экземплярах и повторяли трижды.
https://doi.org/10.1371/journal.pone.0039851.g003
Затем мы оценили влияние мутантов CaM на активность редуктазы eNOS. Цитохром c принимает электроны исключительно из домена FMN и использовался для измерения потока электронов внутри домена редуктазы.Связывание CaM дикого типа с eNOS индуцировало ~ 4-кратное увеличение восстановления цитохрома c . Мутант B 3Q увеличил восстановление цитохрома c до уровня, сопоставимого с СаМ дикого типа (рис. 3В). С другой стороны, снижение цитохрома с , вызванное B 1Q , B 2Q и B 4Q , составило 63%, 67% и 55% от уровня дикого типа, соответственно. Множественные мутанты , то есть B 12Q , B 34Q и B 1234Q , демонстрировали небольшое повышение активности цитохрома с редуктазы или не проявляли его вовсе.Эти результаты демонстрируют, что, за исключением B 3Q , одиночная мутация в сайтах связывания кальция в CaM влияет на активность редуктазы больше, чем активность оксигеназы в eNOS. Более того, полное связывание кальция в N- и C-концевых долях важно для переноса электронов через домен редуктазы eNOS (Рисунок 3B).
Связывание мутантов CaM с пептидом связывания CaM домена eNOS (CBD)
Оценивали способность различных мутантов CaM связывать CBD. Каждый мутант CaM инкубировали с CBD в присутствии 100 мкМ кальция при нескольких различных соотношениях пептид: CaM.Комплексы CBD-CaM и свободный CaM исследовали невосстанавливающим неденатурирующим электрофорезом. Свободные пептиды не были обнаружены, потому что они положительно заряжены при нейтральном pH и, следовательно, будут иметь восходящую подвижность к катоду и не попадут в гель [34], [35]. В присутствии EGTA полосы сдвига подвижности не были обнаружены для дикого типа или любого мутанта CaM (данные не показаны). Комплексы пептид-CaM можно увидеть над полосой свободного CaM в присутствии 100 мкМ кальция. Степень взаимодействия СаМ-пептид оценивают путем измерения ослабления свободных полос СаМ и сдвига подвижности белка СаМ с увеличением концентрации пептида.Как показано на фиг. 4, B 1Q , B 2Q и B 3Q связывали CBD со сродством, аналогичным CaM дикого типа. B 4Q и B 12Q связывали CBD, но связывание не было полным. B 34Q и B 1234Q не связывали CBD. Эти результаты предполагают, что канонический сайт связывания CaM на eNOS взаимодействует с связанной с кальцием C-долей в CaM.
Рисунок 4. Взаимодействие CaM-связывающего домена с каждой конструкцией CaM.
Канонический CaM-связывающий домен eNOS человека (CBD, остатки 491–510) инкубировали с конструкциями CaM (200 пмоль) путем увеличения молярных соотношений пептид: CaM при комнатной температуре в течение 1 ч перед электрофорезом в присутствии 100 мкМ кальций.Образцы анализировали на 18% неденатурирующих гелях и визуализировали с помощью Coomassie Blue R-250. Первая полоса в каждом геле содержит только CaM, , т.е. отношение CBD / CaM равно 0. Остальные отношения CBD / CaM указаны. Обозначены комплексы CBD-CaM и свободный CaM.
https://doi.org/10.1371/journal.pone.0039851.g004
Различная роль N- и C-концевых долей CaM во флюоресценции флавинов eNOS
Поскольку N- и C-концевые доли CaM могут функционировать как независимый домен [33], [36], мы определили, зависит ли преобразование субдомена FMN из экранированного в деэкранированное состояние от связанных с кальцием N- и C- терминальные доли CaM.Поскольку интенсивность флуоресценции флавина пропорциональна степени деэкранирования FMN [37], мы отслеживали изменение флуоресценции флавина eNOS, индуцированное CaM дикого типа, B 12Q или B 34Q в присутствии или в отсутствие кальция. . Как показано на фиг. 5A, CaM дикого типа и мутантные CaM не могли увеличивать флавиновую флуоресценцию eNOS в присутствии 2 мМ EGTA. При добавлении кальция B 34Q увеличивал интенсивность флуоресценции флавина до того же уровня, что и CaM дикого типа.Напротив, B 12Q увеличивал гораздо меньшую интенсивность флуоресценции по сравнению с CaM дикого типа или B 34Q , что указывает на то, что N-концевые EF-руки в его связанной с кальцием форме ответственны за колебания ввода / вывода домена FMN. (Рисунок 5B).
Рис. 5. Флуоресценция флавинов eNOS, обработанных мутантами дикого типа и CaM.
A: Спектры испускания флуоресценции eNOS были получены в растворе, содержащем 25 мМ Трис, pH 7,5, 100 мМ NaCl, 0,1 мМ DTT, ∼6 мкМ eNOS и ∼12 мкМ CaM дикого типа, B 12Q или B 34Q в присутствии 2 мМ EGTA.B: Изменение интенсивности флуоресценции CaM дикого типа, B 12Q или B 34Q регистрировали при добавлении 2,5 мМ CaCl 2 к вышеуказанным смесям. Длину волны возбуждения устанавливали на 455 нм. Данные флуоресценции корректировали путем вычитания СаМ и буферных эффектов. Только eNOS и комплекс eNOS-CaM указаны на каждом графике.
https://doi.org/10.1371/journal.pone.0039851.g005
Трипсинолиз eNOS в присутствии СаМ дикого типа, B12Q или B34Q
Поскольку результаты, представленные выше, предоставили доказательства того, что N- и C-концевые доли CaM в его связанной с кальцием форме играют разные роли во взаимодействии со связывающим доменом CaM и в изменении состояния входа / выхода домена FMN, мы предположили, что связывание из этих двух долей могут вызывать разные конформационные изменения в eNOS.Мы использовали ограниченное расщепление трипсином для обнаружения изменений конформации eNOS, вызванных диким типом, по сравнению с B 12Q или B 34Q . Ограниченный трипсинолиз eNOS проводили в присутствии высоких или низких концентраций кальция.
В отсутствие кальция трипсинолиз eNOS, инкубированных с диким типом, B 12Q или B 34Q , давал идентичный профиль фрагмента с одной основной полосой, имеющей кажущуюся молекулярную массу ~ 56 кДа. N-концевое секвенирование показало, что эта полоса состоит из двух частей N-концевых последовательностей K 29 QGPA (фрагмент-1a на фиг. 6A) и K 498 EVAN (фрагмент-2a на фиг. 6A), что указывает на два расщепления. сайты: (1) между Lys28 и Gln29 и (2) между Lys497 и Glu498.Lys28 находится близко к сайту пальмитоилирования, а Lys497 находится в каноническом CaM-связывающем домене eNOS (фиг. 6C), что отражает то, что без CaM-защиты CaM-связывающий домен eNOS чувствителен к расщеплению трипсином.
Рисунок 6. Ограниченный трипсинолиз eNOS в присутствии CaM дикого типа, B 12Q или B 34Q .
A: Трипсинолиз eNOS (~ 5 мкм) выполняли в присутствии 10 мкМ СаМ дикого типа, B 12Q или B 34Q при 22 ° C в течение 15 минут с 1 мМ EGTA.B: Трипсинолиз проводили в присутствии 100 мкМ CaCl 2 . Дорожка 1: непереваренный eNOS с СаМ дикого типа; полоса 2: трипсинизированные eNOS с СаМ дикого типа; полоса 3: непереваренный eNOS с B 12Q ; полоса 4: трипсинизированная eNOS с B 12Q ; полоса 5: непереваренный eNOS с B 34Q ; полоса 6: трипсинизированная eNOS с B 34Q . Молекулярные массы маркерных белков показаны слева в килодальтонах (кДа). После SDS-PAGE триптические образцы переносили на PVDF-мембраны с использованием буфера CAPS в течение 3 часов при 50 В и 4 ° C.Показаны полосы, вырезанные для N-концевого секвенирования. C: Относительное положение N-концевых последовательностей указанных триптических фрагментов в схематической структуре eNOS. Сайты терминации на С-конце неизвестны, и остаток на С-конце в каждом триптическом фрагменте приблизительно оценивается по кажущейся молекулярной массе. Красная стрелка показывает место расщепления трипсином. Блоки указывают сайты связывания для гема, H 4 B, CaM, FMN, FAD и NADPH. AI и CT относятся к аутоингибиторной петле и С-концевому хвосту соответственно.Обозначены сайты миристоилирования (Myr.) И сайты пальмитоилирования (Palm.).
https://doi.org/10.1371/journal.pone.0039851.g006
В присутствии кальция расщепление трипсином eNOS с CaM дикого типа дало три заметные полосы ∼67 кДа (фрагмент-1b), ∼58 кДа. (фрагмент-2b) и ~ 52 кДа (фрагменты-4b и 5b) (Фиг.6B). N-концевое секвенирование полосы ∼67 кДа выявило сайт расщепления на K 29 QGPA и, судя по кажущемуся размеру, С-концевой триптический сайт, вероятно, находится около Arg630, который расположен в аутоингибиторной (AI) области (рис. 6C, фрагмент-1b), что согласуется с предыдущим сообщением о том, что связывание кальция / CaM индуцирует конформационные изменения eNOS, приводящие к воздействию аутоингибиторных элементов [38].N-концевое секвенирование полосы ∼58 кДа показало сайт расщепления по K 29 QGPA и С-концевой триптический остаток примерно на 549, на основании оценки размера (фрагмент 2b на фиг. 6C). N-концевое секвенирование полосы ∼52 кДа выявило две N-концевые последовательности: (1) фрагмент-4b с N-концевой последовательностью R 722 DIFS и предполагаемым C-концевым концом на остатке 1203, и (2) фрагмент- 5b с N-концевой последовательностью K 517 RVKA и предполагаемым C-концевым концом в остатке 994 (фиг. 6C).
Расщепление трипсином eNOS с помощью B 12Q дало профиль фрагментов, аналогичный профилю CaM дикого типа, за исключением того, что часть N-концевой последовательности фрагмента-3b не была обнаружена в CaM дикого типа, а фрагмент 5b не обнаружен. в B 12Q (Рисунок 6B). N-концевая последовательность фрагмента-3b — это K 498 EVAN, которая находится в каноническом CaM-связывающем домене в eNOS (фиг. 6C). Напротив, профиль фрагментов трипсинолиза eNOS с помощью B 34Q выявил ослабленный фрагмент-1b и усиленный фрагмент-2b и 3b, а также фрагменты 4b и 5b.Данные трипсинолиза дают важную информацию о влиянии CaM на конформацию eNOS: (1) Расщепление eNOS по Lys28 наблюдалось либо в присутствии, либо в отсутствие CaM, что указывает на то, что последовательность этого остатка рядом с сайтом пальмитоилирования находится в зоне воздействия. . ENOS подвергается двойному ацилированию посредством N -миристоилирования по Gly2 и тиопальмитоилирования по Cys15 и Cys26 [39], [40]. Поскольку пальмитоилирование обратимо, вопрос о том, контролирует ли существование регулируемых циклов пальмитоилирования и депальмитоилирования деградацию eNOS, остается предметом дальнейшего исследования; (2) N-концевая последовательность K 498 EVAN (фрагмент-3b) была обнаружена с помощью B 12Q и B 34Q , но не с CaM дикого типа; Интенсивность фрагмента-3b в B 12Q была слабее, чем в B 34Q .Это согласуется с анализом сдвига подвижности геля, в котором B 12Q связывает CaM-связывающий домен, но не так хорошо, как CaM дикого типа; B 34Q не показывает привязки вообще. Результат дополнительно подтверждает, что С-концевой участок в его связанной с кальцием форме может защищать CaM-связывающий домен eNOS от триптического расщепления; (3) связывание eNOS дикого типа, B 12Q или B 34Q аналогичным образом приводило к подверганию R 722 DIFS расщеплению трипсином. Эта последовательность расположена в шарнирной области между доменами FMN и FAD (рис. 6C).Наши предыдущие находки предполагают, что эта область может взаимодействовать с CaM и участвует в катализе eNOS [15]. То, что связывание мутантов дикого типа или CaM, нарушенных в связывании кальция в C- или N-доле, вызвало аналогичное воздействие на шарнирную область, предполагает очень гибкую природу этой области, которая, вероятно, будет служить стержнем для движения FMN домен [41]; (4) Экспозиция K 517 RVKA (фрагмент-5b) со связыванием дикого типа и B 34Q , но не с B 12Q , предполагает, что связывание N-концевой доли CaM в связанной с кальцием форме раскрывает Arg517 для трипсина. пищеварение.eNOS Arg517 аналогичен iNOS Arg536 и nNOS Arg757, которые, как сообщается, образуют как солевой мостик, так и взаимодействие водородных связей с CaM Glu47 и несколькими атомами кислорода основной цепи в субдомене NOS FMN [42], и считаются решающими для контроля взаимодействий субдомена FMN с его окислительно-восстановительные партнеры [43].
Обсуждение
Результаты показывают, что каждый кальций-связывающий сайт на СаМ играет роль в опосредовании СаМ-индуцированного переноса электронов eNOS и каталитической активности. Одиночная мутация каждого сайта связывания приводит к увеличению ЕС50 в ~ 2-3 раза по сравнению с диким типом без существенного влияния на максимальное образование цитруллина.Однако двойная мутация сайтов связывания в C-доле или N-доле CaM резко снижает максимальное образование цитруллина и восстановление цитохрома c , указывая на необходимость специфичного для долей связывания кальция для активности редуктазы eNOS и оксигеназы. Анализ сдвига подвижности геля, измерения флуоресценции флавина и ограниченный трипсинолиз демонстрируют, что N- и C-концевые доли играют особую роль в активации eNOS, , т.е. N-доля в ее связанной с кальцием форме контролирует движение субдомена FMN, в то время как C-доля в своей связанной с кальцием форме связывает область связывающего домена CaM в eNOS.
Взяв вместе результаты нашего текущего исследования, мы обрисовали эти находки в схематической диаграмме, основанной на димерной модели NOS от Даффа [44] и двухлепестковой структуре CaM от Rodney et al. [45], иллюстрируя, как N- и C-концевые доли CaM по-разному регулируют катализ eNOS (Figure 7). В отсутствие кальция апо-СаМ не связан с eNOS, в которой Lys497 чувствителен к расщеплению трипсином; домен FMN, тесно связанный с доменом FAD, находится в заблокированном положении, и поток электронов блокируется (рис. 7A).При связывании с кальцием СаМ дикого типа претерпевает конформационные изменения; Оборачивается вокруг CaM-связывающего домена и, возможно, других CaM-связывающих сайтов в eNOS, при этом две его доли связаны, как это было предложено для множественных CaM-связывающих последовательностей в eNOS (15). Связывание защищает Lys497 от расщепления трипсином и подвергает Arg517 трипсинолизу. Следовательно, домен FMN высвобождается из закрытого положения в открытое, становясь доступным для гемового домена противоположной субъединицы или для экзогенного акцептора электронов, такого как цитохром c (рис. 7B).При двойной мутации N-доли B 12Q CaM привязан к N-концу CaM-связывающего домена eNOS исключительно с помощью связанной с кальцием С-доли. Эта ассоциация защищает Lys497 от триптического расщепления с небольшой индукцией подвижности домена FMN. В этом состоянии домен FMN в основном находится в заблокированном положении, что приводит к значительному ослаблению активности eNOS. (Рисунок 7C). С двойной мутацией C-доли B 34Q , связанная с кальцием N-доля неспособна защитить Lys497 от расщепления трипсином, но подвергает Arg517 трипсинолизу и увеличивает флуоресценцию флавина до того же уровня, что и CaM дикого типа.Это означает, что B 34Q не связывается с СаМ-связывающим доменом, а скорее с областью, прилегающей к СаМ-связывающему домену. В этом состоянии, хотя домен FMN находится в открытом положении, активность оксигеназы и редуктазы eNOS значительно ослаблена, что указывает на то, что освобожденный домен FMN больше не связывается со своими окислительно-восстановительными партнерами, как это обычно происходит (рис. 7D). Это ясно показывает, что связывание между CaM и CaM-связывающим доменом eNOS происходит «голова к хвосту», при этом C-доля CaM связана с N-частью CaM-связывающего домена.Связывание с кальцием, специфичное для долей, имеет решающее значение для CaM для контроля переноса электронов eNOS, а для эффективной активации требуется, чтобы N- и C-концевые доли CaM в его связанной с кальцием форме взаимодействовали с CaM-связывающим доменом, а также с другими CaM-связывающими элементами. в eNOS.
Рис. 7. Схематическое изображение дифференциальной регуляции переноса электронов eNOS N- и C-концевыми долями CaM.
Димерная модель eNOS получена из Daff [44]. Домены , окрашенные в серый цвет, представляют одну субъединицу, а домены , окрашенные в голубой цвет, представляют другую.Блоки указывают сайты связывания для гема, H 4 B, цинка, CaM, FMN, FAD и NADPH. Канонический СаМ-связывающий домен (остатки 491–510) между доменом FMN и доменом гема обозначается как CBD. CaM, показанный как две лопастные структуры N и C, соединенные гибким линкером, адаптирован из ссылки 45; apoCaM отмечен белыми кружками, а связывание кальция обозначено желтыми кружками. A: В отсутствие кальция апо-СаМ не связан с eNOS, в которой Lys497 подвергается расщеплению трипсином; домен FMN заблокирован доменом FAD, и поток электронов заблокирован.B: После связывания с кальцием CaM дикого типа оборачивается вокруг CBD eNOS и, возможно, других сайтов со связанными двумя его долями. Это связывание защищает Lys497 от расщепления трипсином и подвергает Arg517 трипсинолизу. Следовательно, домен FMN высвобождается из домена FAD, становясь доступным для гемового домена противоположной субъединицы. C: При мутации N-доли B 12Q связанная с кальцием C-доля обычно связывается с CBD, в то время как мутированная N-доля не может связывать кальций и не может переносить домен FMN в гемовый домен другой субъединицы, что препятствует перенос электронов, как показано пунктирной линией.D: При мутации C-доли B 34Q C-доля не может связывать CBD, что замедляет перенос электронов.
https://doi.org/10.1371/journal.pone.0039851.g007
Сравнение мутаций кальцийсвязывающего сайта при образовании цитруллина и снижении цитохрома c c в eNOS по сравнению с другими изоформами показывает значительные различия. Как показано в таблице 3, мутация одного кальций-связывающего сайта CaM оказывает меньшее влияние на активацию eNOS, чем на nNOS или iNOS.Примечательно, что мутант B 1Q снижал образование цитруллина eNOS на 25%, но полностью отменял образование цитруллина nNOS [21] и, наоборот, не влиял на образование цитруллина iNOS [22]. С другой стороны, мутант B 3Q , который не влиял на eNOS и вызывал умеренное снижение образования цитруллина nNOS, уменьшал образование цитруллина iNOS более чем на 50%, в то время как B 4Q , который увеличивал образование цитруллина eNOS, вызывал глубокое снижение nNOS, но имел мало влияет на образование цитруллина iNOS.B 2Q не оказывал значительного влияния на образование цитруллина eNOS, но оказывал большое влияние на образование цитруллина nNOS и iNOS. B 3Q селективно снижал образование цитруллина iNOS, тогда как B 4Q снижал образование цитруллина nNOS. Эффект мутации с одним сайтом на восстановление цитохрома с согласуется с действием на образование цитруллина, за исключением B 4Q , который снижает восстановление цитохрома с без значительного влияния на образование цитруллина.Наши данные показывают, что лепестковая мутация сайтов связывания кальция (B 12Q и B 34Q одинаково) серьезно влияет на активность eNOS и nNOS, не влияя на iNOS (23). Эти результаты свидетельствуют о различном влиянии кальций-связывающего статуса CaM на поток электронов и каталитическую активность в eNOS по сравнению с nNOS и iNOS.
B 4Q по сравнению с CaM дикого типа ослаблял снижение цитохрома c на ~ 45%, в то время как он увеличивал образование цитруллина (рисунок 3B vs.3А). Этот дифференциальный эффект является неожиданным, поскольку кристаллографический анализ CaM, связанного с CaM-связывающим доменом eNOS [14] или CaM-связывающим доменом nNOS, а также комплексом субдоменов CaM-iNOS FMN [42], показал взаимодействие CaM с каноническим CaM-связывающим доменом. сайт всех трех изоформ NOS в антипараллельной ориентации, в которой сайт B 4Q (E140 CaM) взаимодействует с N-концевой частью CaM-связывающего домена, расположенной поблизости от домена оксигеназы NOS. Неясно, как мутация этого сайта влияет только на активность редуктазы без снижения активности оксигеназы.Тем не менее, результаты подтверждают мнение о том, что СаМ активирует редуктазу и оксигеназу по разным механизмам [46].
Недавняя кристаллическая структура CaM-связанного субдомена iNOS FMN [42] показала, что Glu47, расположенный в N-концевой доле CaM, участвует в мостиковом взаимодействии с доменом iNOS FMN, которое помогает трансдукции эффектов связанного CaM. Авторы предположили, что аналогичное мостиковое взаимодействие субдоменов CaM-FMN должно присутствовать в ферментах nNOS и eNOS. Tejero et al.Позже доказал, что это мостовое соединение, по-видимому, контролирует перемещение субдоменов nNOS FMN [43]. Здесь мы предполагаем, что это мостиковое взаимодействие в первую очередь контролируется специфическим для долей связыванием кальция в CaM. Наши данные по флуоресценции флавина демонстрируют, что B 34Q способен индуцировать интенсивность флуоресценции eNOS до того же уровня, что и CaM дикого типа, предполагая взаимодействие между CaM Glu47 и субдоменом eNOS FMN, который присутствует в выходном состоянии. Однако B 34Q практически не увеличивает активность оксигеназы и редуктазы eNOS, возможно, из-за дефектного связывания с СаМ-связывающим доменом.Эти данные предполагают, что прикрепление связанной с кальцием C-концевой доли CaM к CaM-связывающему домену eNOS запускает FMN-домен в более подходящей ориентации для взаимодействия с его окислительно-восстановительными партнерами. Поскольку B 12Q индуцировал гораздо меньшее увеличение флуоресценции флавина, чем CaM дикого типа, возможно, что мостиковое взаимодействие между CaM Glu47 и субдоменом eNOS FMN требует N-концевой доли CaM в его связанной с кальцием форме. Из этих результатов мы заключаем, что связанные с кальцием C-доли и N-доли CaM взаимодействуют с eNOS в разных областях скоординированным образом, что необходимо для эффективного переноса электронов между различными окислительно-восстановительными центрами в eNOS.
Материалы и методы
Материалы
L- [2,3,4,5 -3 H] аргинин (NET1123250UC) был получен от PerkinElmer Inc. (6R) -5,6,7,8, -Тетрагидро-L-биоптерин (H 4 B) был приобретен у Research Biochemical International. Реагент белкового красителя Bradford и электрофоретические химикаты были продуктами Bio-Rad. Рестрикционные ферменты были от New Englands Biolabs. СаМ-агароза, AG 50W-X8 (катионообменная смола), НАДФН и другие реагенты были получены от Sigma.Клетка насекомого Sf21 была получена от Clontech (номер заказа 631411).
Мутагенез
кДНК CaM человека была любезно предоставлена доктором Эмануэлем Э. Стрелером. Серия одноточечных мутантов CaM человека с заменами Glu (E) на Gln (Q) в положениях z для координации кальция в каждой EF-руке была создана с помощью сайт-направленного мутагенеза и обозначена как B 1Q (E31Q), B 2Q (E67Q), B 3Q (E104Q) и B 4Q (E140Q) соответственно.Множественные точечные мутанты, неспособные связывать кальций ни с N-концевой долей (B 12Q ; мутации E31Q и E67Q), ни с С-концевой долей (B 34Q ; мутации E104Q и E140Q), так и с обеих долей (B 1234Q ). ; E31Q, E67Q, E104Q и E140Q). Основная стратегия основанного на ПЦР мутагенеза была описана ранее [47]. Праймеры, использованные для мутации, и остатки, измененные для каждого мутанта, перечислены в таблице 1. Мутированные остатки глутамина (CAA или CAG) выделены жирным шрифтом.Все праймеры были синтезированы MdBio Inc. (Тайбэй, Тайвань). СаМ дикого типа и мутантные клонировали в вектор pT7-7 через сайты NdeI и HindIII. Правильные мутации были подтверждены службой секвенирования в основном учреждении NHRI в Мяоли, Тайвань. Секвенированные гены CaM были такими же, как в Genbank под номером доступа J04046.
Экспрессия и очистка CaM дикого типа и мутант
BL21 (DE3), трансформированный каждой конструкцией CaM, выращивали при 37 ° C в среде LB с добавлением 100 мкг / мл ампициллина.Культуры индуцировали 0,3 мМ изопропи-β-D-тиогалактопиранозидом, когда OD 600 достигали 0,8–1,2, и собирали после экспрессии в течение ночи при комнатной температуре. Очистку различных СаМ проводили, как описано ранее [15], [48]. Концентрация СаМ определялась либо методом Брэдфорда [49], либо спектрофотометрически с использованием 277 нм = 2560 M -1 см -1 .
Выражение и очистка eNOS
кДНК eNOS человека вставляли в сайт Eco RI вектора переноса pVL1392, который использовали для создания рекомбинантных вирусов в системе Sf21 / бакуловирус.Из-за низкой биосинтетической способности гема клеток Sf21, дополнительный хлорид гема (4 мкг / мл) был добавлен в культуральную среду через 18 ч после инфицирования для увеличения содержания гема в экспрессируемой eNOS. Клетки собирали через 60 ч после инфицирования, суспендировали в буфере, содержащем 25 мМ Трис-HCl, pH 7,5, 0,2 мМ дитиотреитола (DTT), 0,1 мМ ЭДТА, 0,1 мМ EGTA, 1 мкМ пепстатина А, 1 мкМ лейпептина, 1 мкМ антипаина, 1 мкМ фенилметилсульфонила. фторид и 10% глицерин, а затем трижды обрабатывали ультразвуком в течение 20 с, центрифугировали дважды при 30,000 × g в течение 20 минут при 4 ° C.Супернатант загружали в аффинную колонку 2 ‘, 5′-ADP-Sepharose (1,5 × 5 см), белки элюировали буфером, содержащим 25 мМ Трис-HCl, pH 7,5, 0,1 мМ DTT, 20 мМ 2’ и 3 ‘. Смесь -АМР, 0,5 М NaCl и 10% глицерин. Элюат разбавляли 4-объемным буфером, содержащим 25 мМ Трис-HCl, pH 7,5, 0,1 мМ DTT, 100 мкМ CaCl 2 и 10% глицерин, и наносили на колонку с CaM-агарозой (1,5 × 5 см) перед предварительной обработкой. уравновешивают буфером, содержащим 25 мМ трис-HCl, pH 7,5, 0,1 мМ DTT, 100 мкМ CaCl 2 , 100 мМ NaCl и 10% глицерин (буфер A).Колонку промывали буфером А и элюировали буфером, содержащим 25 мМ Трис-HCl, pH 7,5, 0,1 мМ DTT, 2 мМ EGTA, 0,5 М NaCl и 10% глицерин. Элюат концентрировали с помощью Centriprep-50 (Millipore) и диализовали против буфера, содержащего 25 мМ трис-HCl, pH 7,5, 0,1 мМ DTT, 100 мМ NaCl и 10% глицерин.
Устойчивое состояние активности eNOS
АктивностьeNOS определяли путем измерения превращения L- [3H] аргинина в L- [3H] цитруллин, как описано ранее [4] с небольшими изменениями.Реакционная смесь, содержащая 25 мМ трис-HCl, pH 7,5, 0,1 мМ DTT, 100 нМ eNOS, 300 мкМ CaCl 2 , 0,2 мМ EDTA, 100 мкМ β-NADPH, 10 мкМ H 4 B, 20 мкМ L- аргинин, 10% глицерин и 1 мкКи L- [ 3 H] аргинина инкубировали при 37 ° C в течение 4 минут либо с фиксированным количеством (0,5 мкМ), либо с различными концентрациями каждой конструкции CaM. Восстановление цитохрома с определяли при комнатной температуре в реакционной смеси, содержащей 25 мМ трис-HCl, pH 7,5, 100 мМ NaCl, 10% глицерин, 50 мкМ цитохром с с ,0.5 мкМ CaM, 100 мкМ CaCl 2 и 100 мкМ β-НАДФН. Реакцию инициировали добавлением 50 нМ eNOS и контролировали при 550 нм в спектрофотометре GE geneQuant100. Активность определяли с использованием Cred-oxi 21 мМ -1 см -1 .
Анализ изменения подвижности геля
Канонический СаМ-связывающий пептид (CBD), TRKKTFKEVANAVKISASLM (остатки eNOS 491–510) был синтезирован компанией Peptide 2, Inc. (Шантильи, Вирджиния). Каждую конструкцию CaM (200 пмоль) инкубировали с CBD при увеличивающихся молярных отношениях в течение 1 ч при комнатной температуре в буфере (10 мкл), содержащем 20 мМ Трис, pH 7.5 либо 100 мкМ CaCl 2 , либо 1 мМ EGTA. Образец подвергали 18% -ному неденатурирующему, невосстанавливающему PAGE [50], который проводили при 30 мА в условиях с высоким содержанием кальция (100 мкМ свободного кальция в гелевых буферах) или в условиях с низким содержанием кальция (1 мМ EGTA в гелевых буферах). Связывание CBD с CaM визуализировали окрашиванием кумасси синим R-250.
Флуоресцентная спектроскопия
Стационарную флуоресценцию флавина измеряли в растворе, содержащем 25 мМ Трис, pH 7,5, 100 мМ NaCl, 0.1 мМ DTT, 2 мМ EGTA, 10% глицерин и ~ 6 мкМ eNOS в отсутствие или в присутствии ~ 12 мкМ конструкций CaM с использованием флуоресцентного спектрофотометра VARIAN CARY Eclipse. Для экспериментов использовалась кварцевая кювета объемом 1 мл с длиной оптического пути 1 см. Общее изменение объема сохранялось менее 0,3%, и во время измерения образцы поддерживались при комнатной температуре. Влияние связывания СаМ дикого типа и мутантного СаМ на флуоресценцию флавина eNOS отслеживали при добавлении 2,5 мМ CaCl 2 .Белок возбуждали при 455 нм, и спектры испускания регистрировали в диапазоне от 460 до 700 нм в течение 5 минут для каждого образца. Спектры излучения были скорректированы с учетом фона из-за эффектов буфера и CaM.
Трипсинолиз eNOS
Ограниченный протеолиз проводили в смеси, содержащей 5 мкМ рекомбинантной eNOS, очищенной из клеток Sf21, 50 мМ Tris, pH 7,5, 100 мМ NaCl, 10 мкМ дикого типа или мутантного CaM, и либо в присутствии 100 мкМ CaCl. 2 или 1 мМ EGTA.Образцы предварительно инкубировали при комнатной температуре в течение 15 минут, и протеолиз инициировали добавлением 0,1 нг трипсина золота (Promega) на пмоль eNOS. После инкубации при комнатной температуре в течение 15 мин реакцию останавливали кипячением с равным объемом буфера для загрузки геля 2X SDS. Протеолитические продукты были разделены с помощью SDS-PAGE с градиентом 8–16% [50] и визуализированы путем окрашивания кумасси синим R250.
Анализ N-концевой последовательности
После SDS-PAGE триптические образцы переносили на мембраны из поливинилидендифторида (PVDF) с использованием буфера CAPS в течение 3 часов при 50 В и 4 ° C.Полосы окрашивали красным Понсо и вырезали для секвенирования. Секвенирование выполняли на белковом секвенаторе Applied Biosystems Model 494 в Mission Biotech Inc. (Тайбэй, Тайвань).
Благодарности
кДНК CaM человека была подарком д-ра Эмануэля Э. Стрелера.
Вклад авторов
Задумал и спроектировал эксперименты: PFC. Проведены эксперименты: PRW. Проанализированы данные: PFC PRW CCK. Предоставленные реагенты / материалы / инструменты анализа: CCK JYL.Написал статью: PFC KKW SFY.
Ссылки
- 1. Рафиков Р., Фонсека Ф.В., Кумар С., Пардо Д., Дарра С. и др. (2011) активация eNOS и функция NO: структурные мотивы, ответственные за посттрансляционный контроль активности эндотелиальной синтазы оксида азота. J Endocrinol 210: 271–284.
- 2. Дудзинский Д.М., Мишель Т. (2007) История жизни eNOS: партнеры и пути. Сердечно-сосудистые исследования 75: 247–260.
- 3. Schmidt HH, Wilke P, Evers B, Bohme E (1989) Ферментативное образование оксидов азота из L-аргинина в цитозоле бычьего мозга.Biochem Biophys Res Commun 165: 813–819.
- 4. Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR, et al. (1991) Клонированная и экспрессированная синтаза оксида азота по структуре напоминает цитохром Р-450 редуктазу. Nature 351: 714–718.
- 5. Stuehr DJ, Cho HJ, Kwon NS, Weise M, Nathan C (1991) Очистка и характеристика цитокин-индуцированной синтазы оксида азота макрофагов: флавопротеин, содержащий FAD и FMN. Proc Natl Acad Sci U S A 88: 7773–7777.
- 6. Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, et al. (1992) Клонирование и характеристика индуцибельной синтазы оксида азота из макрофагов мыши. Science 256: 225–228.
- 7. Pollock JS, Förstermann U, Mitchell JA, Warner TD, Schmidt HH и др. (1991) Очистка и характеристика синтазы релаксирующего фактора эндотелия в виде частиц из культивируемых и природных эндотелиальных клеток аорты крупного рогатого скота. Proc Natl Acad Sci U S A 88: 10480–10484.
- 8. Sheta EA, McMillan K, Masters BS (1994) Доказательства бидоменной структуры конститутивной синтазы оксида азота мозжечка. J. Biol Chem. 269: 15147–15153.
- 9. Ghosh DK, Stuehr DJ (1995) Макрофагальная NO-синтаза: характеристика изолированных доменов оксигеназы и редуктазы выявляет прямое взаимодействие субъединиц. Биохимия 34: 801–807.
- 10. Чен П.Ф., Цай А.Л., Берка В., Ву К.К. (1996) Эндотелиальная синтаза оксида азота.Доказательства структуры бидомена и успешного восстановления каталитической активности двух отдельных доменов, генерируемых системой экспрессии бакуловируса. J Biol Chem 271: 14631–14635.
- 11. Abu-Soud HM, Yoho LL, Stuehr DJ (1994) Кальмодулин контролирует нейрональную синтазу оксида азота по двойному механизму. Активация внутри- и междоменного переноса электрона. J Biol Chem 269: 32047–32050.
- 12. Сиддханта У., Преста А., Фан BC, Волан Д., Руссо Д.Л. и др.(1998) Обмен доменов в индуцибельной синтазе оксида азота. Перенос электронов происходит между группами флавина и гема, расположенными на соседних субъединицах в димере. J Biol Chem 273: 18950–18958.
- 13. Сагами И., Дафф С., Шимицу Т. (2001) Внутрисубъединичный и межсубъединичный перенос электронов в нейрональной синтазе оксида азота: влияние кальмодулина на катализ гетеродимеров. J Biol Chem 276: 30036–30042.
- 14. Aoyagi M, Arvai AS, Tainer JA, Getzoff ED (2003) Структурная основа связывания эндотелиальной синтазы оксида азота с кальмодулином.Embo J 22: 766–775.
- 15. Chen PF, Wu KK (2009) Два синтетических пептида, соответствующие проксимальному гем-связывающему домену и домену CD1 эндотелиальной синтазы оксида азота человека, ингибируют активность оксигеназы, взаимодействуя с CaM. Arch Biochem Biophys 486: 132–140.
- 16. Бабу Ю.С., Багг С.Е., Кук В.Дж. (1988) Структура кальмодулина, очищенная при разрешении 2,2 A. J Mol Biol 204: 191–204.
- 17. Wang CL (1985) Заметка о связывании кальция с кальмодулином.Biochem Biophys Res Commun. 130: 426–430.
- 18. Meador WE, Means AR, Quiocho FA (1992) Распознавание целевого фермента кальмодулином: 2.4 Структура комплекса кальмодулин-пептид. Science 257: 1251–1255.
- 19. Cho HJ, Xie QW, Calaycay J, Mumford RA, Swiderek KM, et al. (1992) Кальмодулин представляет собой субъединицу синтазы оксида азота из макрофагов. J Exp Med 176: 599–604.
- 20. Бредт Д.С., Снайдер С.Х. (1990) Выделение синтетазы оксида азота, фермента, требующего кальмодулина.Proc Natl Acad Sci U S A 87: 682–685.
- 21. Stevens-Truss R, Beckingham K, Marletta MA (1997) Сайты связывания кальция кальмодулина и перенос электронов нейрональной синтазой оксида азота. Биохимия 36: 12337–12345.
- 22. Грибовская И., Браунлоу К.С., Деннис С.Дж., Роско А.Дж., Марлетта М.А. и др. (2005) Кальций-связывающие сайты кальмодулина и перенос электронов индуцибельной синтазой оксида азота. Биохимия 44: 7593–7601.
- 23. Spratt DE, Taiakina V, Guillemette JG (2007) Кальций-дефицитное связывание кальмодулина и активация нейрональных и индуцибельных синтаз оксида азота.Biochim Biophys Acta 1774: 1351–1358.
- 24. Govers R, Rabelink TJ (2001) Клеточная регуляция эндотелиальной синтазы оксида азота. Am J Physiol Renal Physiol 280: F193 – F206.
- 25. Shifman JM, Choi MH, Mihalas S, Mayo SL, Kennedy MB (2006) Кальций / кальмодулин-зависимая протеинкиназа II (CaMKII) активируется кальмодулином с двумя связанными кальциями. Proc Natl Acad Sci USA. 103: 13968–13973.
- 26. Craig DH, Chapman SK, Daff S (2002) Кальмодулин активирует перенос электронов через нейрональный домен редуктазы синтазы оксида азота, высвобождая NADPH-зависимый конформационный замок.J Biol Chem 277: 33987–33994.
- 27. Ghosh DK, Salerno JC (2003) Синтазы оксида азота: доменная структура и выравнивание в функции и контроле ферментов. Front Biosci 8: 193–209.
- 28. Гарсин Э.Д., Брунс К.М., Ллойд С.Дж., Хосфилд Д.Д., Тисо М. и др. (2004) Структурные основы изоферментной регуляции переноса электронов в синтазе оксида азота. J Biol Chem 279: 37918–37927.
- 29. Roman LJ, Masters BS (2006) Перенос электронов нейрональной синтазой оксида азота регулируется согласованным взаимодействием кальмодулина и двух внутренних регуляторных элементов.J Biol Chem 281: 23111–23118.
- 30. Ли Х., Дас А., Сибхату Х., Джамал Дж., Слигар С.Г. и др. (2008) Изучение свойств переноса электронов нейрональной синтазы оксида азота путем изменения окислительно-восстановительного потенциала FMN. J Biol Chem 283: 34762–34772.
- 31. Илаган Р.П., Тисо М., Конас Д.В., Хеманн С., Дурра Д. и др. (2008) Различия в конформационном равновесии различают катализ эндотелиальными и нейрональными флавопротеинами синтазы оксида азота. J Biol Chem 283: 19603–19615.
- 32. Lee A, Zhou H, Scheuer T, Catterall WA (2003) Молекулярные детерминанты Ca (2 +) / кальмодулин-зависимой регуляции каналов Ca (v) 2.1. Proc Natl Acad Sci USA 100: 16059–16064.
- 33. Джама А.М., Габриэль Дж., Аль-Нагар А.Дж., Мартин С., Баиг С.З. и др. (2011) Доли-специфические функции кальций-кальмодулина в активации кальций-кальмодулин-зависимой протеинкиназы II. J Biol Chem 276: 30794-30802.
- 34. Romanin C, Gamsjaeger R, Kahr H, Schaufler D, Carlson O, et al.(2000) Ca (2+) сенсоры Ca (2+) канала L-типа. FEBS lett 487: 301–306.
- 35. Тан В., Холлинг ДБ, Блэк Диджей, Пейт П, Чжан Дж. З. и др. (2003) Апокальмодулин и сайты связывания кальмодулина кальмодулина в канале CaV1.2. Biophys J 85: 1538–1547.
- 36. Persechini A, McMillans K, Leakey P (1994) Активация активности киназы легкой цепи миозина и синтазы оксида азота фрагментами кальмодулина. J. Biol Chem. 269: 16148–16154.
- 37. Adak S, Ghosh S, Abu-Soud HM, Stuehr DJ (1999) Роль кислотных остатков кластера 1 редуктазного домена в нейрональной синтазе оксида азота.Характеристика фермента FMN-FREE. J Biol Chem 274: 22313–22320.
- 38. Салерно Дж. К., Харрис Д. Е., Иризарри К., Патель Б., Моралес А. Дж. И др. (1997) Аутоингибиторный контрольный элемент определяет регулируемые кальцием изоформы синтазы оксида азота. J Biol Chem 272: 29769–29777.
- 39. Liu J, Sessa WC (1994) Идентификация ковалентно связанной аминоконцевой миристиновой кислоты в эндотелиальной синтазе оксида азота. J. Biol Chem. 269: 11691–11694.
- 40.Liu J, Garcia-Cardeiia G, Sessa WC (1995) Биосинтез и пальмитоилирование эндотелиальной синтазы оксида азота: мутагенез сайтов пальмитоилирования, цистеинов-15 и / или -26, выступает против перемещения фермента, вызванного депальмитоилированием. Биохимия 34: 12333–12340.
- 41. Хак М.М., Кустубх П., Теджеро Дж., Аулак К.С., Фадлалла М.А. и др. (2007) Соединительный шарнир подавляет активность эндотелиальной синтазы оксида азота. Proc Natl Acad Sci U S A 104: 9254–9259.
- 42. Xia C, Misra I, Iyanagi T, Kim JJ (2009) Регулирование междоменных взаимодействий кальмодулином в индуцибельной синтазе оксида азота. J Biol Chem 284: 30708-30717.
- 43. Tejero J, Haque1 MM, Durra D, Stuehr DJ (2010) Мостиковое взаимодействие позволяет кальмодулину активировать NO-синтазу через бимодальный механизм. J Biol Chem 285: 25941–25949.
- 44. Daff S (2010) NO-синтаза: структуры и механизмы. Оксид азота 23: 1–11.
- 45. Родни Г.Г., Крол Дж., Уильямс Б., Бекингем К., Гамильтон С.Л. (2001) Карбоксиконцевые участки связывания кальция кальмодулина контролируют переключение кальмодулина с активатора на ингибитор RYR1. Биохимия 40: 12430–12435.
- 46. Ньюман Э., Спратт Д.Е., Мошер Дж., Чейн Б., Монтгомери Х.Дж. и др. (2004) Дифференциальная активация изоферментов синтазы оксида азота химерами кальмодулин-тропонин С. J Biol Chem 279: 33547–33557.
- 47. Chen PF, Tsai AL, Wu KK (1994) Цистеин 184 эндотелиальной синтазы оксида азота участвует в координации гема и каталитической активности.J Biol Chem 269: 25062–25066.
- 48. Pitt GS, Zűhlke RD, Hudmon A, Schlman H, Reuter H, et al. (2001) Молекулярные основы связывания кальмодулина и кальций-зависимой инактивации кальциевых каналов L-типа. J Biol Chem 276: 30794-30802.
- 49. Брэдфорд М.М. (1976) Быстрый и чувствительный метод количественного определения количества белка в микрограммах, использующий принцип связывания белок-краситель. Анальная биохимия 72: 248–254.
- 50. Laemmli UK (1970) Расщепление структурных белков во время сборки головки бактериофага Т4.Природа 227: 680–685.
Активация секретагогической независимой секреции желудочной кислоты посредством стимуляции эндотелиальной синтазы оксида азота у крыс — FullText — Cellular Physiology and Biochemistry 2017, Vol. 44, № 4
Абстрактные
Предпосылки / цели: L-аргинин является важным медиатором деления клеток, заживления ран и иммунной функции. Он может быть преобразован синтазой оксида азота (NOS) в оксид азота (NO), важную клеточную сигнальную молекулу.Недавние исследования в нашей лаборатории демонстрируют специфические эффекты воздействия L-аргинина (10 мМ) на секрецию желудочного сока париетальными клетками крыс. Методы: Исследования проводились с изолированными желудочными железами и pH-чувствительным красителем BCECF-AM +/- L-аргинин для изучения его влияния на секрецию кислоты. Гидрат натриевой соли диэтиламина NONOate прямого донора NO также использовали при мониторинге внутриклеточного pH. Также использовали специфический ингибитор внутриклеточного сигнального каскада NO ODQ. Результаты: Мы обнаружили, что экструзия протонов желудка активировалась применением L-аргинина (10 мМ), в отдельной серии, когда добавлялись L-аргинин (10 мМ) + L-NAME (30 мкМ), секреция кислоты отсутствовала. Добавление NO-донора гидрата натриевой соли диэтиламина NONOate (10 мкМ) также вызывало секрецию кислоты. При добавлении селективного sGC-ингибитора ODQ с NONOate секреция кислоты не наблюдалась. Заключение: Мы пришли к выводу, что L-аргинин является новым стимулятором секреции, который может опосредовать секрецию желудочной кислоты.Кроме того, потребление L-аргинина вызывает прямую активацию АТФазы H + , K + и усиление экструзии протонов из париетальных клеток, что приводит к повышенному риску заболеваний, связанных с кислотой. Путь NO / sGC / cGMP никогда ранее не описывался как возможный внутриклеточный механизм активации АТФазы H + , K + и представляет собой совершенно новое научное открытие. Более того, наши исследования демонстрируют новую роль L-NAME в эффективном устранении индуцированной NOS секреции кислоты и, таким образом, в снижении риска язвенной болезни, индуцируемой L-аргинином.
© 2017 Автор (ы). Опубликовано S. Karger AG, Базель
Введение
L-аргинин относится к полузаменимым аминокислотам [1] и является единственным эндогенным субстратом, несущим азот, для синтеза эндотелиального оксида азота (NO) синтазой оксида азота (NOS) [2-4]. Наряду с синтезом NO L-аргинин служит субстратом для ферментативной трансформации агматина, креатина и мочевины в организме человека [4, 5].Как единственный предшественник и донор азота для оксида азота (NO), L-аргинин играет центральную роль в форме посредника в нескольких физиологических и патофизиологических процессах [6-9]. Таким образом, L-аргинин можно рассматривать как сигнальную молекулу, которая отвечает за поддержание основных клеточных функций, например. синтез белков, гибель и рост клеток [4, 10, 11]. В качестве полузаменимой аминокислоты L-аргинин может поступать из эндогенных и экзогенных источников. Почки могут поддерживать базальный синтез L-аргинина de-novo [4, 12], но особенно во время высокой потребности в L-аргинине, такой как рост, воспаление или процессы заживления ран, потребление богатой L-аргинином пищи имеет решающее значение для поддержания адекватного предложения [4, 13, 14].Следовательно, рекомендуется диетическое потребление L-аргинина в количестве 1–3 г / сут с пищей [4, 15]. Импорт гидрофобной аминокислоты через биологические мембраны происходит через множество транспортных систем внутри липидного бислоя. Захват аргинина париетальными клетками желудочной железы может быть Na + -независимым или Na + -зависимым. Переносчик катионных аминокислот (CAT), переносчик гетеро (ди) аминокислот (HAT) и переносчик пептидов (PEPT) ответственны за внутриклеточное поглощение L-аргинина.Изоформы CAT имеют широкую экспрессию и повсеместно представлены [16, 17]. Они работают, не зависимо от Na + , и в равной степени переносят другие аминокислоты в клетку, например орнитин и лизин [18]. HAT, особенно изоформа rBAT / b 0, + , ответственны за трансэпителиальный катионный обмен аминокислот между клетками, а PEPT1 и PEPT2 являются H + -зависимыми переносчиками ди- и трипептидов [19, 20]. Для получения дополнительной информации о транспортерах L-аргинина, пожалуйста, обратитесь к элегантному обзору Closs et al.(Ссылка 4) «Плазменные мембранные переносчики аргинина». Как только L-аргинин попадает в клетку, NOS превращает его в NO и побочный продукт L-цитруллин [21]. Существуют различные типы NOS, которые различаются по местоположению и функциям: eNOS (NOS-3, эндотелиальный), nNOS (NOS-1, нейрональный) и iNOS (NOS-2, индуцибельный) [22, 23]. Сообщалось о наличии митохондриальной БДУ, но ученые не смогли идентифицировать структуру и функцию до настоящего времени [23, 24]. Эндотелиальная синтаза оксида азота (eNOS) отвечает за регуляцию артериального давления и ангиогенез посредством внутриклеточных сигнальных трансдукций [22, 25, 26], тогда как нейрональная NOS (nNOS) участвует в нейротрансмиссии, процессах обучения и синаптической пластичности [3, 25 ].Изоформы eNOS и nNOS действуют через кальций-зависимый путь и регулируют нейрональные и сосудистые процессы посредством связывания кальмодулина [23]. Третья изоформа системы NOS, индуцируемая NOS (iNOS), играет важную роль во врожденных иммунных ответах и антипатогенных процессах во время инфекций вирусами и бактериями [25, 27, 28] и модулирует развитие опухоли [22, 25, 28] . Система iNOS не зависит от кальция и производит большое количество газа и радикалов соответственно, образуя цитотоксическое микроокружение внутри клетки, которое можно обнаружить во время инфекции и развития опухоли [7].NO считается одной из важнейших внутриклеточных сигнальных молекул [21]. Он отвечает за регуляцию кровотока и приводит к расширению сосудов [7, 23, 29] и ингибированию агрегации тромбоцитов [23, 30]. Кроме того, NO является важным триггером сердечного развития во время эмбриогенеза [31]. Активация внутриклеточного рецептора NO-GC через NO приводит к увеличению содержания растворимой гуанилилциклазы (рГЦ) и, в дальнейшем, к накоплению циклического гуанозинмонофосфата (цГМФ) [31, 32]. Мы предполагаем, что секреция желудочного сока в париетальных клетках, стимулированная L-аргинином, происходит через сигнальный путь NO / NO-GC / cGMP и может увеличивать риск всех связанных с кислотой заболеваний, таких как язвенная болезнь (GUD), неэрозивный рефлюкс. болезнь (НЭРБ), гастроэзофагеальная рефлюксная болезнь (ГЭРБ) вплоть до развития злокачественных новообразований желудка и пищевода.Таким образом, L-аргинин может рассматриваться как потенциальный возбудитель этих желудочно-кишечных заболеваний, а чрезмерное потребление L-аргинина может привести к серьезным нарушениям для желудка. Другими хорошо известными причинами являются нестероидные противовоспалительные препараты (НПВП), никотин, алкоголь, кофеин, стресс и инфекции [33–36]. Поскольку распространенность гастроэзофагеальных заболеваний в США постоянно растет [37], новые данные о возбудителях заболевания представляют большой интерес.
Настоящее исследование фокусируется на внутриклеточных изменениях скорости восстановления pH после применения L-аргинина и предоставляет подробную информацию о механизме и внутриклеточных характеристиках секреции кислоты желудочного сока, вызванной L-аргинином.Мы выбрали L-NAME в качестве неселективного ингибитора NOS и смогли предотвратить желудочную секрецию путем ингибирования стимулированного L-аргинином сигнального пути NO / NO-GC / cGMP в париетальных клетках.
Материалы и методы
Животные и подготовка желудка
Мы использовали самцов крыс Sprague-Dawley массой 250-400 г (Charles River Laboratory), содержащихся в помещениях с регулируемым климатом и влажностью, которым давали стандартный корм с неограниченный доступ к воде 24 часа. С животными обращались в соответствии с гуманной практикой ухода за животными, установленной Комитетом по уходу и использованию животных в Йельском университете.Животных лишали пищи на 12-16 часов с неограниченным доступом к воде перед тем, как их усыпили передозировкой изофлурана. Мы сделали лапаротомию, удалили желудок и продольным разрезом отделили тело желудка. После выделения тела его промывали ледяным забуференным раствором Рингера HEPES для удаления остаточных частиц пищи и разрезали на квадратные срезы ткани 0,2 см. Процедура, в том числе резекция желудка, была выполнена быстро, и небольшие срезы тканей хранили на льду в чашке Петри, наполненной ледяным буферизованным раствором Рингера HEPES, чтобы гарантировать жизнеспособность и функцию клеток желудочных желез [38].
Ручная диссекция и изоляция желудочных желез
Отдельные срезы тела переносили на диссекционный микроскоп и изолировали отдельные железы с помощью техники ручной диссекции, как описано ранее [39, 40]. Вкратце, корпус разрезали на квадраты 2 см x 2 см, а затем каждый квадрат дополнительно разрезали на отдельные секции 1 см x 1 см. Мышечный слой отделяли от среза в бане с ледяным буферным раствором Рингера при 4 ° C. Отдельные железы желудка вырезали из этих срезов и затем переносили в холодном растворе Рингера в перфузионную камеру.Желудочным железам позволяли прилипать к покровным стеклам, покрытым 0,5 мкл биологического адгезива Corning CellTak (Collaborative Research, Массачусетс, США), широко используемого и хорошо известного тканевого адгезива на основе полифенольных белков, который может прочно связывать как неорганические, так и органические вещества. поверхности в водной среде [41]. Соответственно, покровные стекла были прикреплены к перфузионной камере, как описано ранее [37, 39]. Перед нанесением красителя на покрытые покровные стекла переносили как минимум 4-6 желез.
Цифровая визуализация для измерения внутриклеточного pH
Изолированные желудочные железы инкубировали в растворе Рингера, забуференном HEPES и содержащем 10 мкМ pH-чувствительного красителя BCECF-AM (2´, 7´) -bis- (2). -карбоксиэтил) -5- (и-6) -карбоксифлуресцин-ацетометиловый эфир (Santa Cruz Biotechnology, TX, USA) в течение 20 минут.После инкубации красителя перфузионная камера была помещена на предметный столик инвертированного микроскопа (Olympus IX50) и промыта раствором Рингера с буфером HEPES в течение 5 минут для удаления остаточного неэтерифицированного красителя. Во время этой фазы промывки температура камеры и перфузата была повышена до 37oC. В камере контролировали термостат, и до конца исследования поддерживали температуру 37 ° C. Использовался режим эпифлуоресценции с объективом x40, и нагруженный BCECF сальник возбуждали на длинах волн 490 нм ± 10 нм и 440 нм ± 10 нм.На каждую железу выделяли минимум 8 интересующих областей, за которыми одновременно наблюдали в течение всей экспериментальной процедуры. Результирующие флуоресцентные сигналы BCECF успешно отслеживались на длине волны 530 ± 10 нм каждые 10 секунд с помощью камеры устройства с усиленной зарядовой связью. Отдельные изображения и значения интенсивности были записаны вместе с данными излучения, показывающими соотношение 490/440 для измерений интенсивности в реальном времени [37]. В конце каждого экспериментального цикла клетки калибровали с помощью метода калибровки High K + / нигерицин для преобразования всех ратиометрических данных и расчета внутриклеточного pH из данных эмиссии, как описано в предыдущих исследованиях [42].
Восстановление внутриклеточного pH (pH i ) от кислотной нагрузки демонстрирует скорость экструзии протонов отдельными париетальными клетками после кислотно-протонной нагрузки с использованием метода предымпульса NH 4 Cl [39]. Таким образом, клетки были подвергнуты суперфузии с раствором с буфером HEPES, содержащим 40 мМ NH 4 Cl, для загрузки клеток ионами водорода. Затем париетальные клетки промывали свободным раствором Na + , чтобы исключить активацию Na-H-обменника (NHE) и улавливание протонов в цитозоле, вызывая сильное падение pH –.Затем клетки промывали HEPES и, наконец, калибровочным раствором HighK + , чтобы зафиксировать внутриклеточный pH до 7,0. Скорости восстановления были выражены в dpH i / dT (DpH / мин) для всех исследований после преобразования данных о соотношении с использованием уравнения Хендерсона-Хассельбаха, как описано ранее [43, 44]. Наблюдаемая скорость восстановления внутриклеточного pH (pH –) представляет собой экструзию протонов париетальными клетками. Интенсивность восстановления внутриклеточного pH и рассчитывалась исходя из одного и того же начального исходного pH, чтобы исключить изменение индивидуальной внутриклеточной буферной способности клеток в различных экспериментальных условиях.Скорость восстановления выражена в DpH / мин.
Химические вещества и реагенты
Температура и pH всех растворов (см. Таблицу 1) были доведены до 37 ° C и 7,4, за исключением калибровочного раствора High K + , который титровали до 7,0. Конечная осмоляльность всех растворов составляла 300 мОсм. Все химические вещества, включая L-аргинин, L-NAME (метиловый эфир NG-нитро-1-аргинина), карбахол, ODQ (1H- [1, 2,4] оксадиазоло [4, 3-a] хиноксалин-1-он) и гидрат натриевой соли NONOate диэтиламина были получены от Sigma Aldrich (St.Луис, Миссури, США) и Дж. Бейкер (Филлипсбург, Нью-Джерси, США).
Таблица 1.
Решения. Концентрации выражены в миллимолях
Статистический анализ
Программное обеспечение Graphpad / Prism 7.0 использовалось для анализа различий в скоростях восстановления pH и . Мы выполнили непарный параметрический t-критерий Стьюдента, чтобы продемонстрировать существенные различия в скоростях восстановления pH и . Значения DpH / мин представлены как среднее ± SEM. Все результаты с p <0,05 считались достоверными.
Результаты
Чтобы изучить быстрое клеточное действие L-аргинина на секрецию кислоты, были проведены исследования с использованием изолированного препарата желудочной железы. В отсутствие стимуляции (контроль) наблюдалась низкая базальная скорость восстановления pH — (0,0003711 ± 0,0006969 ΔpHi / мин).
В первой серии экспериментов желудочные железы, подвергнутые слитию с растворами, содержащими L-аргинин (10 мМ), были способны вызывать секреторную стимуляцию, приводящую к повышению скорости восстановления pH — (0.02999 ± 0,002718 DpH / мин) (рис.1).
Рис. 1.
L-аргинин активирует секрецию кислоты желудочного сока. Гистограмма суммирует эффект 10 мМ L-аргинина на покоящиеся париетальные клетки (0,02999 ± 0,002718 pH / мин) (n = 146). Изолированные желудочные железы крыс были загружены чувствительным к pH красителем BCECF и возбуждены при 490 нм ± 10 нм и 440 нм ± 10 нм. Скорость восстановления pH и рассчитывалась по наклону в отсутствие Na + после кислотной нагрузки с использованием метода предымпульса NH 4 Cl [39].Контроль показывает низкий базальный отток протонов из покоящейся париетальной клетки в отсутствие стимулирующих агентов (0,0003711 ± 0,0006969 ΔpHi / мин) (n = 66).
В экспериментах с L-аргинином можно заметить очень значительную разницу в экструзии протонов по сравнению с контрольными условиями (p <0,0001).
В отдельной серии экспериментов мы использовали растворы, содержащие комбинацию L-NAME (30 мкМ) и L-аргинина (10 мМ). Наши результаты показывают, что мы можем достичь значительного снижения индуцированной L-аргинином секреции желудочной кислоты в присутствии неселективного ингибитора NOS L-NAME по сравнению с зависимой от L-аргинина секрецией желудочной кислоты в отсутствие L-NAME (0.002185 ± 0,0004337 DpH / мин против 0,02999 ± 0,002718DpH / мин, p <0,0001) (рис. 2).
Рис. 2.
L-NAME может ингибировать индуцированную L-аргинином секрецию желудочной кислоты. После добавления в наши растворы неселективного ингибитора NOS L-NAME результаты демонстрируют низкий базальный отток протонов из покоящейся париетальной клетки в присутствии стимулирующего агента L-аргинина (0,002185 ± 0,0004337 ΔpH / мин) (n = 179) .
В другой серии экспериментов мы индуцировали стимулирующий эффект вытеснения протонов с помощью классического стимулятора секреции карбахола (200 мкМ) и получили степень восстановления pH i , равную 0.03291 ± 0,002645 DpH / мин. Чтобы убедиться, что экструзия протонов карбахолом чувствительна к L-NAME, мы добавили L-NAME (30 мкМ) в растворы, содержащие карбахол. Экструзия протонов, чувствительная к карбахолу, не ингибировалась L-NAME, и скорости извлечения не показали существенной разницы в pHi по сравнению с зависимой от карбахола секрецией желудочной кислоты в отсутствие L-NAME (0,03291 ± 0,0026450 DpH / мин по сравнению с 0,03362 ± 0,0040260 DpH. / мин, p = нс) (рис.3).
Рис. 3.
Гистограмма сравнивает действие классического средства, стимулирующего секрецию карбахола, на желудочные железы крыс (0.03291 ± 0,0026450 ΔpH / мин) (n = 168) к комбинации карбахола и L-NAME вместе (0,03362 ± 0,0040260 ΔpH / мин) (n = 104). Наши результаты демонстрируют, что индуцированная карбахолом секреция желудочной кислоты не может быть подавлена ингибитором NOS L-NAME. Мы пришли к выводу, что L-аргинин и карбахол различаются по своему внутриклеточному пути, ведущему к сборке и окончательной активации АТФазы H + , K + .
Мы сравнили скорости восстановления pH и L-NAME (30 мкМ) + L-аргинин (10 мМ) с L-NAME (30 мкМ) + карбахол (200 мкМ), продемонстрировав весьма значительную разницу в секреции кислоты желудочного сока (0.002185 ± 0,0004337 против 0,03362 ± 0,0040260 DpH / мин, p = 0,0001) (рис. 4).
Рис. 4.
Столбиковая диаграмма, подчеркивающая разницу между L-аргинином и карбахолом: секреция желудочной кислоты, индуцированная L-аргинином, может подавляться L-NAME (0,002185 ± 0,0004337 ΔpH / мин) (n = 179), тогда как стимуляция секрецию кислоты классическим стимулятором секреции карбахолом невозможно предотвратить применением L-NAME (0,03362 ± 0,0040260 ΔpH / мин) (n = 104). Таким образом, оба агента различаются по своему внутриклеточному пути индукции секреции.Карбахол приводит к внутриклеточному увеличению уровней цАМФ с использованием мускариновых рецепторов, что индуцирует вставку и активацию АТФаз H + , K + , тогда как секреция желудочной кислоты, индуцированная L-аргинином, действует через путь NO / sGC / cGMP, блокируемый ингибитором eNOS L-NAME.
В последней серии экспериментов мы подтвердили нашу гипотезу о том, что L-аргинин действует через внутриклеточный путь NO в железах желудка. Поэтому мы добавили к нашим растворам 10 мкМ гидрата натриевой соли диэтиламина NONOate прямого донора NO [45].Применение донора NO привело к увеличению экструзии H + из париетальной клетки (0,03093 ± 0,002054 DpH / мин). Добавляя мощный и селективный ингибитор чувствительной к оксиду азота ингибитора гуанилилциклазы ODQ (1H- [1, 2,4] оксадиазоло [4, 3-a] хиноксалин-1-он) (10 мкм) к нашему гидрату натриевой соли NONOate диэтиламина, содержащему растворов мы смогли свести к минимуму влияние на активацию АТФаз H + , K + до базального уровня (0,00009813 ± 0,0006929 DpH / min).Интересно, что мы сохранили почти идентичные результаты для протонной экструзии с комбинацией растворов, содержащих ODQ (10 мкМ) и L-аргинин (10 мМ) (0,001077 ± 0,00048 DpH / мин) (рис. 5).
Рис. 5.
Гистограмма, показывающая влияние прямого донора NO. Мы нанесли NO-донорный диэтиламин NONOate натрия гидрат (NONO) 10 мкМ на наш раствор, и результаты демонстрируют увеличение секреции кислоты желудочного сока (0,03093 ± 0,002054 ΔpH / мин) (n = 183). Добавляя sGC-ингибитор ODQ к нашим растворам, содержащим NONOate, мы смогли полностью устранить этот эффект до уровня базального оттока протонов (0.00009813 ± 0,0006929 ΔpH / мин) (n = 140). Кроме того, мы объединили растворы, содержащие L-аргинин, с ODQ и достигли почти аналогичного эффекта (0,001077 ± 0,00048 ΔpH / мин) (n = 185). Таким образом, мы доказываем, что секреция желудочной кислоты, стимулированная L-аргинином, зависит от внутриклеточного пути NO / sGC.
Наши результаты свидетельствуют о том, что наблюдения связаны с образованием NO, соответственно опосредованным цГМФ и рГЦ, поскольку секреция желудочной кислоты, индуцированная L-аргинином, может подавляться проницаемым для клеток и селективным ингибитором рГЦ ODQ, что приводит к эффективному ингибированию циклического ГМФ. производство.
Обсуждение
Наши результаты демонстрируют, что L-аргинин является мощным активатором NOS, который может индуцировать секрецию кислоты через активацию АТФазы H + , K + в железах покоя или может также работать в активированных железах через независимый путь. (Рис. 6). Наши данные убедительно свидетельствуют о том, что повышенные уровни L-аргинина в крови после еды могут вызывать непрерывную секрецию кислоты париетальными клетками даже в отсутствие классических стимуляторов секреции и нормальных модуляторов секреции кислоты, стимулирующей постсекретацию (рис.7). Когда мы провели отдельную серию исследований в присутствии неселективного ингибитора NOS L-NAME, мы смогли предотвратить секрецию кислоты, индуцированную L-аргинином. Этот результат предполагает, что существует дополнительная петля обратной связи, связанная с генерацией и разложением NO в желудочной железе, которая модулирует секрецию кислоты в отсутствие обычных стимуляторов секреции.
Рис. 6.
Модель париетальной клетки желудочной железы. Модель дает обзор переносчиков и рецепторов ионов, а также нейроэндокринной регуляции секреции кислоты желудочного сока.Компоненты концентрированной соляной кислоты секретируются протонными насосами (H + , K + АТФаз) и хлоридными каналами в просвет желудка. L-аргинин проникает в париетальную клетку через транспортные системы CAT, PEPT и HAT и ферментативно превращается во внутриклеточную сигнальную молекулу NO посредством eNOS. Увеличение NO следует за увеличением рГЦ, что вызывает повышение внутриклеточных уровней цГМФ и в конечном итоге приводит к встраиванию и активации АТФазы H + , K + на апикальной мембране париетальной клетки.Внутриклеточное увеличение цАМФ (через гистамин) или Ca2 + (через гистамин, ацетилхолин, Ca2 + или гастрин) после стимуляции базолатеральных рецепторов оказывает такое же влияние на введение и активацию H + , K . + АТФаз, таких как цГМФ. Прямая стимуляция нейронов происходит за счет ацетилхолина из ENS, тогда как гормональная регуляция париетальных клеток зависит от вицинальных, секретирующих гистамин ECL-клеток и от G-клеток, расположенных в антральной оболочке, которые секретируют гастрин.Гастрин также стимулирует секрецию гистамина в ECL-клетках и приводит к увеличению секреции гистамина, что описывается как «ось гистамин-гастрин». Соматостатин, секретируемый D-клетками, снижает внутриклеточные уровни цАМФ и останавливает вставку и активацию АТФаз H + , K + .
Рис. 7.
Схема экспериментального курса. График объясняет индивидуальный эксперимент с L-аргинином (сводка всех экспериментов с L-аргинином на рис. 1), частота растворов и смесь растворов с лекарством.Мы использовали стандартный HEPES, 40 мМ NH 4 Cl, 0Na и High K + (см. Состав растворов в таблице 1) в сочетании с 10 мМ L-Arg. Растворы меняли каждые 5 минут.
Это исследование демонстрирует новый эффект L-аргинина на париетальные клетки желудка, его эффективность как стимулятора секреции и влияние на секрецию желудочного сока. Более того, мы подчеркиваем важность NOS в L-аргинин-зависимом внутриклеточном сигнальном пути, поскольку мы можем отменить индуцированную L-аргинином секрецию желудочной кислоты путем применения неселективного ингибитора NOS L-NAME.Мы предлагаем eNOS в качестве важного посредника, участвующего в этом процессе, благодаря селективным активаторам и ингибиторам, которые мы использовали в нашем исследовании. Кроме того, основываясь на нашем многолетнем опыте использования изолированного препарата желудочной железы и предыдущих публикациях, у нас никогда не было никаких доказательств того, что рассеченная единственная железа имеет активную нейронную цепь.
Исследования Price et al. показали, что конститутивная форма NOS представлена в слизистой оболочке желудка [46]. Конститутивная форма является кальций / кальмодулин-зависимой и включает эндотелиальную (eNOS) и нейрональную (nNOS) изоформы [46].Экспрессия nNOS в париетальных клетках желудка крысы была подтверждена Премаранте с помощью гистохимии NADPH-диафоразы, иммуноокрашивания nNOS, вестерн-блоттинга и ОТ-ПЦР и опубликована в 2001 году [47]. Однако методика, использованная для этого исследования, отличается от нашей: они использовали иммуно-препарированные железы, тогда как мы использовали вручную препарированные железы. Экспрессия изоформ изменяется при патологических условиях, например канцерогенез или инфекция желудка H. pylori. Экспрессия конститутивных изоформ NOS изменялась в зависимости от стадий злокачественных новообразований и варьировала от снижения до почти полной потери до сверхэкспрессии ферментов.Интересно, что iNOS экспрессируется в тканях кардии и дна желудка, хотя в физиологических условиях он не представлен [48].
Связь между секрецией кислоты желудочного сока и путем NO / NO-GC / cGMP, кроме того, подчеркивается нашими экспериментами с гидратом натриевой соли NONOate диэтиламина и ODQ с ингибитором sGC. Применение гидрата натриевой соли NONOate диэтиламина, являющегося прямым донором NO, увеличивает секрецию кислоты в желудке и подтверждает нашу гипотезу о том, что секреция кислоты желудочного сока, индуцированная L-аргинином, является производной от NO.Эксперименты с ингибитором рГЦ ODQ демонстрируют его способность подавлять секрецию кислоты желудочного сока, индуцированную как NONOate, так и L-аргинином. Таким образом, ODQ может стать потенциально новым средством против желудочно-кишечных заболеваний, связанных с кислотой.
Выражение признательности
Проект был профинансирован Фондом хирургических исследований Чарльза Осе хирургии Йельского университета.
AMK отвечал за проведение экспериментов, обсуждение данных, написание рукописи, построение рисунков, интерпретацию данных, статистический анализ.А.Л. отвечал за конструктивную рецензию окончательной рукописи. JPG задумал ответственный за экспериментальный дизайн, получение финансирования для исследования, интерпретацию данных и рецензирование окончательной рукописи.
Заявление о раскрытии информации
Авторы заявляют, что у них нет конфликта интересов в отношении содержания этой статьи.
Список литературы
- Вернер А., Аманн Е., Шнитциус В., Хабермайер А., Лакнер-Минден С., Лейхтнер Н., Рупп Дж., Клосс Е. И., Мундер М. Индуцированный транспорт аргинина через переносчик катионных аминокислот-1 необходим для пролиферации Т-клеток человека.Eur J Immunol 2016; 46: 92-103.
- Фигероа А., Вонг А., Хайме С.Дж., Гонсалес Ю.Ю.: Влияние добавок L-цитруллина и арбуза на функцию сосудов и работоспособность. Curr Opin Clin Nutr Metab Care 2017; 20: 92-98.
- Mungrue IN, Bredt DS: nNOS вкратце: значение для мозга и мускулов.J Cell Sci 2004; 117: 2627-2629.
- Клосс Э.И., Саймон А., Векони Н., Ротманн А.: Плазменные мембранные переносчики аргинина. The J Nutr 2004; 134: 2752S-2759S; обсуждение 2765С-2767С.
- Моррис С.М., мл.: Ферменты метаболизма аргинина. J Nutr 2004; 134: 2743S-2747S; обсуждение 2765С-2767С.
- Albaugh VL, Pinzon-Guzman C, Barbul A: Двойные роли аргинина как онконутриента и иммунонутриента. J Surg Oncol 2017; 115: 273-280.
- Ваннини Ф., Кашфи К., Нат Н: двойная роль iNOS в развитии рака.Редокс Биол 2015; 6: 334-343.
- Куинн А.С., Петрос А.Дж., Валланс P: Оксид азота: эндогенный газ. Br J Anaesth 1995; 74: 443-451.
- Zhang N, Diao Y, Hua R, Wang J, Han S, Li J, Yin Y: пути, опосредованные оксидом азота, и его роль в дегенеративных заболеваниях.Front Biosci (Landmark Ed) 2017; 22: 824-834.
- Эль-Гаяр С., Тюринг-Нахлер Х., Пфайльшифтер Дж., Роллингхофф М., Богдан С.: Трансляционный контроль индуцибельной синтазы оксида азота с помощью IL-13 и доступности аргинина в воспалительных макрофагах. J Immunol 2003; 171: 4561-4568.
- Ли Дж., Рю Х., Ферранте Р. Дж., Моррис С. М., мл., Ратан Р. Р.: Трансляционный контроль индуцибельной экспрессии синтазы оксида азота аргинином может объяснить парадокс аргинина. Proc Natl Acad Sci U S A 2003; 100: 4843-4848.
- Jonker R, Deutz NE, Erbland ML, Anderson PJ, Engelen MP: Изменения метаболизма аргинина в организме при хронической обструктивной болезни легких.Am J Clin Nutr 2016; 103: 1458-1464.
- Скаглиа Ф., Брунетти-Пьерри Н., Клеппе С., Марини Дж., Картер С., Гарлик П., Джахур Ф., О’Брайен В., Ли Б.: Клинические последствия дефицита ферментов цикла мочевины и потенциальные связи с метаболизмом аргинина и оксида азота. J Nutr 2004; 134: 2775S-2782S; обсуждение 2796С-2797С.
- King NE, Rothenberg ME, Zimmermann N: Аргинин при астме и воспалении легких. J Nutr 2004; 134: 2830S-2836S; обсуждение 2853S.
- Мори М., Гото Т: ферменты метаболизма аргинина, оксид азота и инфекция.J Nutr 2004; 134: 2820S-2825S; обсуждение 2853S.
- MacLeod CL: Регулирование экспрессии гена переносчика катионных аминокислот (CAT). Biochem Soc Trans 1996; 24: 846-852.
- Closs EI: Экспрессия, регуляция и функция белков-носителей для катионных аминокислот.Curr Opin Nephrol Hypertens 2002; 11: 99-107.
- Белый MF: транспорт катионных аминокислот через плазматическую мембрану клеток млекопитающих. Biochim Biophys Acta 1985; 822: 355-374.
- Herrera-Ruiz D, Knipp GT: Текущие взгляды на установленные и предполагаемые переносчики олигопептидов млекопитающих.J Pharm Sci 2003; 92: 691-714.
- Дэниел Х., Коттра Г.: Семейство котранспортеров протонных олигопептидов SLC15 в физиологии и фармакологии. Pflugers Arch 2004; 447: 610-618.
- Poulos TL, Li H: Структурная основа для селективного ингибирования изоформ в синтазе оксида азота.Acc Chem Res 2013; 46: 390-398.
- Санхуэса С., Араос Дж., Наранхо Л., Баррос Е., Субиабре М., Толедо Ф, Гутьеррес Дж., Кьярелло Д. И., Пардо Ф, Лейва А., Собревия Л.: Оксид азота и модуляция pH при гинекологическом раке. J Cell Mol Med 2016; 20: 2223-2230.
- Коне BC: Молекулярная биология натрийуретических пептидов и синтаз оксида азота.Cardiovasc Res 2001; 51: 429-441.
- Гафурифар П., Рихтер С. Активность синтазы оксида азота в митохондриях. FEBS Lett 1997; 418: 291-296.
- Брунель А., Ланг Дж., Кутюр М., Буше Дж. Л., Дорле П., Сантолини Дж .: Активация кислорода в NO-синтазах: доказательства прямой роли субстрата.FEBS Open Bio 2016; 6: 386-397.
- Sessa WC: краткий обзор eNOS. J Cell Sci 2004; 117: 2427-2429.
- Богдан Ц .: Оксид азота и иммунный ответ.Nat Immunol 2001; 2: 907-916.
- Lowenstein CJ, Padalko E: iNOS (NOS2) с первого взгляда. J Cell Sci 2004; 117: 2865-2867.
- Козаева Л.П., Городецкая Е.А., Рууге Е.К., Каленикова Е.И., Медведев О.С.: Благоприятное влияние инъекции кофермента Q10 на вызванное оксидом азота расширение аорты крысы.Eur J Pharmacol 2016; 794: 15-19.
- Радомски М.В., Палмер Р.М., Монкада С. Путь L-аргинина / оксида азота, присутствующий в тромбоцитах человека, регулирует агрегацию. Proc Natl Acad Sci U S A 1990; 87: 5193-5197.
- Spinelli V, Vona A, Corti F, Diolaiuti L, Zanardelli M, Sartiani L, Failli P: роль оксида азота, синтазы оксида азота, растворимой гуанилилциклазы и cGMP-зависимой протеинкиназы I в развитии стволовых клеток мыши.Стволовые клетки Int 2016; 2016: 2868323.
- Koesling D, Mergia E, Russwurm M: Физиологические функции NO-чувствительных изоформ гуанилилциклазы. Curr Med Chem 2016; 23: 2653-2665.
- Malfertheiner P, Chan FK, McColl KE: Язвенная болезнь.Ланцет 2009; 374: 1449-1461.
- Локк Г.Р., 3-й, Талли Н.Дж., Фетт С.Л., Зинсмайстер А.Р., Мелтон Л.Дж., 3-й: Факторы риска, связанные с симптомами гастроэзофагеального рефлюкса. Am J Med 1999; 106: 642-649.
- Dent J, El-Serag HB, Wallander MA, Johansson S: Эпидемиология гастроэзофагеальной рефлюксной болезни: систематический обзор.Gut 2005; 54: 710-717.
- Ferstl FS, Kitay AM, Trattnig RM, Alsaihati A, Geibel JP: Зависимый от секретагогии и независимый транспорт форм гидратации цинка в париетальных клетках крыс. Pflugers Arch 2016; 468: 1877–1883.
- Кирхгоф П., Сократ Т., Сидани С., Даффи А., Брейдтхардт Т., Гроб С., Виль CT, Беглингер С., Эртли Д., Гейбель Дж. П.: соли цинка обеспечивают новое, длительное и быстрое ингибирование секреции желудочной кислоты.Am J Gastroenterol 2011; 106: 62-70.
- Busque SM, Kerstetter JE, Geibel JP, Insogna K: аминокислоты L-типа стимулируют секрецию кислоты желудочного сока путем активации рецептора, чувствительного к кальцию, в париетальных клетках. Am J Physiol Gastrointest Liver Physiol 2005; 289: G664-669.
- Гейбель Дж. П., Вагнер К. А., Кароппо Р., Куреши И., Глёкнер Дж., Мануэлидис Л., Кирхгоф П., Радебольд К. Шрам, связанный с рецептором двухвалентных ионов желудка, является модулятором секреции желудочной кислоты.J Biol Chem 2001; 276: 39549-39552.
- Boron WF, Waisbren SJ, Modlin IM, Geibel JP: Уникальный барьер проницаемости апикальной поверхности париетальных и главных клеток в изолированных перфузируемых желудочных железах. J Exp Biol 1994; 196: 347-360.
- Sahai S, Wilkerson M, Zaske AM, Olson SD, Cox CS, Triolo F: рентабельный метод иммобилизации гидратированных образцов мягких тканей для атомно-силовой микроскопии.Биотехники 2016; 61: 206-209.
- Paradiso AM, Negulescu PA, Machen TE: Na + -H + и Cl (-) — OH- (HCO3-) обмен в желудочных железах. Am J Physiol 1986; 250: G524-534.
- Боярский G, Ganz MB, Sterzel RB, Boron WF: регуляция pH в отдельных клубочковых мезангиальных клетках.II. Na + -зависимые и -независимые Cl (-) — HCO3- обменники. Am J Physiol 1988; 255: C857-869.
- Боярский G, Ganz MB, Sterzel RB, Boron WF: регуляция pH в отдельных клубочковых мезангиальных клетках. I. Экструзия кислоты в отсутствие и в присутствии HCO3 Am J Physiol 1988; 255: C844-856.
- Wang C, Kemp-Harper BK, Kocan M, Ang SY, Hewitson TD, Samuel CS: Антифиброзные действия релаксина опосредуются NO-sGC-cGMP-зависимым путем в почечных миофибробластах in vitro и усиливаются NO донором , Диэтиламин NONOate. Front Pharmacol 2016; 7: 91.
- Прайс К.Дж., Хэнсон П.Дж., Уиттл Б.Дж.: Локализация конститутивных изоформ синтазы оксида азота в слизистой оболочке желез желудка крысы.Cell Tissue Res 1996; 285: 157-163.
- Premaratne S, Xue C, McCarty JM, Zaki M, McCuen RW, Johns RA, Schepp W, Neu B, Lippman R, Melone PD, Schubert ML: Нейрональная синтаза оксида азота: экспрессия в париетальных клетках крыс. Am J Physiol Gastrointest Liver Physiol 2001; 280: G308-313.
- Rajnakova A, Goh PM, Chan ST, Ngoi SS, Alponat A, Moochhala S: Экспрессия дифференциальных изоформ синтазы оксида азота в нормальной слизистой оболочке желудка человека и ткани рака желудка. Канцерогенез 1997; 18: 1841-1845.
Автор Контакты
Джон П.Geibel
Отделение хирургии Йельской школы медицины,
BML 265, 310 Cedar St., New Haven (USA)
Тел. 203 737 4152, электронная почта [email protected]
Подробности статьи / публикации
Предварительный просмотр первой страницы
Получено: 26 апреля 2017 г.
Принято: 10 ноября 2017 г.
Опубликовано онлайн: 6 декабря 2017 г.
Дата выпуска: декабрь 2017 г.
Количество страниц для печати: 10
Количество рисунков: 7
Количество столов: 1
ISSN: 1015-8987 (печатный)
eISSN: 1421-9778 (онлайн)
Для дополнительной информации: https: // www.karger.com/CPB
Лицензия открытого доступа / Дозировка лекарства / Отказ от ответственности
Эта статья находится под международной лицензией Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 (CC BY-NC-ND). Использование и распространение в коммерческих целях, а также любое распространение измененных материалов требует письменного разрешения. Дозировка лекарств: авторы и издатель приложили все усилия, чтобы гарантировать, что выбор и дозировка лекарств, указанные в этом тексте, соответствуют текущим рекомендациям и практике на момент публикации.Однако ввиду продолжающихся исследований, изменений в правительственных постановлениях и постоянного потока информации, касающейся лекарственной терапии и реакций на них, читателю настоятельно рекомендуется проверять листок-вкладыш для каждого препарата на предмет любых изменений показаний и дозировки, а также дополнительных предупреждений. и меры предосторожности. Это особенно важно, когда рекомендованным агентом является новое и / или редко применяемое лекарство. Отказ от ответственности: утверждения, мнения и данные, содержащиеся в этой публикации, принадлежат исключительно отдельным авторам и соавторам, а не издателям и редакторам.Появление в публикации рекламы и / или ссылок на продукты не является гарантией, одобрением или одобрением рекламируемых продуктов или услуг или их эффективности, качества или безопасности. Издатель и редактор (-ы) не несут ответственности за любой ущерб, причиненный людям или имуществу в результате любых идей, методов, инструкций или продуктов, упомянутых в контенте или рекламе.
Эндотелиальная синтаза оксида азота: потенциальная терапевтическая мишень при цереброваскулярных заболеваниях | Молекулярный мозг
Цереброваскулярные заболевания — это различные сосудистые заболевания мозгового кровообращения.Нарушение подачи артериального кислорода в мозг приводит к цереброваскулярным заболеваниям, таким как транзиторная ишемическая атака, инсульт, субарахноидальное кровоизлияние и сосудистая деменция. Многие механизмы способствуют сложному патофизиологическому процессу. Цереброваскулярные заболевания стали основными причинами длительной инвалидности и смертности во всем мире.
В настоящее время лечение цереброваскулярных заболеваний включает тромболитическую терапию, препараты против агрегации тромбоцитов, антикоагулянты и нейропротекторы.Однако все эти методы лечения имеют серьезные ограничения, включая короткое время лечения, побочные эффекты, осложнения, длительное лечение и большие расходы. Следовательно, цереброваскулярные заболевания являются не только серьезным медицинским бременем, но также тяжелым экономическим и социальным бременем. Это бремя стимулировало неустанные усилия по предотвращению и лечению цереброваскулярных заболеваний и, в частности, по определению роли цереброваскулярного эндотелия в патогенезе цереброваскулярных заболеваний. Цереброваскулярный эндотелий, как сосудистый барьер, напрямую контактирующий с кровью, сначала повреждается сосудистыми факторами риска, а затем формирует различные цереброваскулярные заболевания.Интересно, что эндотелиальные клетки микрососудов головного мозга (BMEC), известные как крупнейший эндокринный, паракринный и метаболический орган в организме, могут генерировать и выделять множество вазоактивных веществ. К ним относятся оксид азота (NO) и эндотелин-1 (ЕТ-1), которые поддерживают цереброваскулярный гомеостаз [1]. Кроме того, цереброваскулярный эндотелий играет решающую роль в регуляции церебрального кровотока (CBF). Частично это происходит за счет синтеза NO, основного вазодилататора, из L-аргинина путем каталитической реакции ферментной эндотелиальной синтазы оксида азота (eNOS).Адекватный CBF — одно из основных условий нормальной работы мозга. Продукция эндотелиального NO в основном зависит от активности eNOS; следовательно, не следует игнорировать активацию eNOS в эндотелии сосудов головного мозга. Недавнее исследование показало, что у пациентов, подвергшихся воздействию сосудистых факторов риска, эндотелий-зависимая дисфункция релаксации выявлялась до каких-либо морфологических изменений на стенке сосуда, что означает, что эндотелий сосудов повреждается в первую очередь. Что еще более важно, различные факторы риска цереброваскулярных заболеваний, включая гипертензию, гиперхолестеринемию, диабет, старение, курение и ожирение, могут нарушать сокращение и расширение цереброваскулярного эндотелия [2].Следовательно, eNOS как важный регулятор функций цереброваскулярного эндотелия следует признать потенциальной терапевтической мишенью для профилактики и лечения цереброваскулярных заболеваний.
Церебральный кровоток и неврологическая функция
Мозг потребляет 20% энергии и питательных веществ организма. Петля базилярной артерии соединяется с общей сонной артерией и охватывает почти весь головной мозг многочисленными ветвями (передняя средняя и задняя мозговые артерии).Ветви простираются от мягкой мембраны к белому веществу, составляющему цереброваскулярную систему. Эта система играет незаменимую роль в поддержке мозга, обеспечивая его кислородом и питательными веществами. В нормальных физиологических условиях мозг может поддерживать адекватный CBF через свои сосудистые механизмы ауторегуляции, чтобы поддерживать энергетические потребности своих клеточных компонентов. Однако многие патологические состояния, включая церебральную ишемию, травму, инсульт и вазоспазм после субарахноидального кровоизлияния (САК), вызывают аномальный CBF, впоследствии вызывая различные нарушения головного мозга.Нарушения церебральной гемодинамики, такие как изменения местного перфузионного давления и целостности сосудов, являются важными патофизиологическими элементами при ишемическом инсульте. Это может привести к дальнейшему повреждению нейронов или глиальных клеток помимо начального ишемического повреждения, вызванного гипо- или гиперперфузией [3]. Существующие данные свидетельствуют о том, что цереброваскулярная эндотелиальная дисфункция, вызванная церебральной ишемией и вызванным аноксией вазоспазмом и / или тромбозом, дополнительно снижает CBF. Постоянное снижение CBF может постепенно вызывать нарушения обучения и памяти, а также когнитивную дисфункцию, что приводит к развитию деменции [4–6].Таким образом, регуляция CBF тесно связана с неврологической функцией. Расширение кровеносных сосудов — важный способ улучшить CBF. Эндотелиальные клетки сосудов регулируют расширение сосудов путем синтеза и секреции цитокинов, таких как PGI 2 и NO. Исследования показали, что эндотелиальный NO, образующийся в эндотелии сосудов головного мозга, является одной из наиболее важных сигнальных молекул механизмов ауторегуляции CBF [7, 8].
Эндотелиальная оксид-синтаза в механизмах регуляции мозгового кровообращения
Как упоминалось выше, мозг наделен вазорегуляторными механизмами, которые гарантируют, что он получает достаточно крови, чтобы поддерживать расход энергии.Эндотелиальный NO является одной из важнейших сигнальных молекул в этих механизмах (рис. 1) [7, 8]. Он участвует в сохранении и поддержании микроциркуляции головного мозга, подавлении агрегации тромбоцитов, адгезии и миграции лейкоцитов, а также в снижении пролиферации гладких мышц. NO играет ключевую роль в регуляции цереброваскулярных эффектов за счет увеличения или уменьшения содержания кислорода, а также повышения содержания углекислого газа, монооксида углерода и ауторегуляции цереброваскулярной системы [9, 10]. Общепризнано, что потеря эндотелиального NO является центральным механизмом в патогенезе эндотелиальной дисфункции.Как в церебральных, так и в периферических сосудистых сетях снижение доступности эндотелиального NO приведет к серьезным пагубным изменениям сосудистых функций. К ним относятся сужение сосудов, повышение артериального давления, пролиферация гладкомышечных клеток сосудов (VSMC), агрегация тромбоцитов, адгезия лейкоцитов и воспаление. Снижение доступности эндотелиального NO играет важную роль в инициации и прогрессировании сосудистых заболеваний, таких как атеросклероз. Следовательно, сохранение эндотелиальной продукции NO является важной стратегией профилактики цереброваскулярных заболеваний [11].Однако различные сосудистые факторы риска могут прямо или косвенно снижать продукцию NO эндотелием. Окислительный стресс, вызванный чрезмерной продукцией активных форм кислорода (АФК) и, прежде всего, супероксид-анионов, считается наиболее важным механизмом снижения эндотелиального NO. С одной стороны, сосудистые факторы риска способствуют усилению активности НАДФН-оксидазы, образованию супероксид-анионов, химической инактивации NO и образованию сильного окислителя, пероксинитрита [12]. С другой стороны, окислительный стресс, вызванный повышенным образованием пероксинитрита, может окислять тетрагидробиоптерин (Bh5), важный сопутствующий фактор, необходимый для активности eNOS.Если концентрация Bh5 становится субоптимальной, димер eNOS будет отделяться от других мономеров eNOS, что приводит к снижению продукции эндотелиального NO и увеличению продукции супероксид-анионов и пероксинитрита [13]. Сообщается, что снижение поступления кислорода в артериальную кровь (гипоксия) увеличивает исходный уровень CBF у людей [14]. Таким образом, NO играет важную роль как в сигнальном событии, так и в ответе CBF [15]. Напротив, неселективное ингибирование NOS устраняет реакцию увеличения CBF на гипоксию у крыс [16].Отсюда следует, что сигнальный путь NO играет центральную роль в ответе CBF на эти изменения, и эта дисфункция пути постишемии вызывает нарушение регуляции этих нормальных физиологических ответов. Недавние исследования регуляции CBF эндотелиальным NO предоставили новое понимание физиологического контроля цереброваскулярных функций и патологических механизмов неврологических заболеваний. Они также предоставляют новые подсказки для разработки новых терапевтических стратегий при церебральной дисфункции [9].Действительно, eNOS уделялось даже больше внимания, чем NO, из-за нестабильности NO и механизмов регуляции eNOS продукции NO.
Рис. 1Синтаза оксида эндотелия (eNOS) и ее роль в механизмах регуляции мозгового кровотока (CBF). eNOS активируется ацетилхолином (ACh), брадикинином, напряжением сдвига и т. д., а затем катализирует L-аргинин с образованием NO. Он превращается в клетки гладких мышц сосудов, вступает в реакцию с гуанилатциклазой (ГК) и способствует превращению гуанозинтрифосфата (ГТФ) в циклический гуанозинмонофосфат (цГМФ), что приводит к расслаблению гладких мышц сосудов и увеличению CBF на
.Эндотелиальная синтаза оксида азота и цереброваскулярные заболевания
Функция eNOS
Врожденная активность eNOS определяет предрасположенность к травмам, поскольку она обеспечивает защиту от вторичного повреждения нейронов.Нарушение активности eNOS из-за SAH, черепно-мозговой травмы (TBI) и ишемического инсульта вовлечено во многие клеточные механизмы повреждения нейронов [17].
Отсроченная ишемия головного мозга (DCI) — одна из основных причин заболеваемости и смертности после САК. Недавнее исследование продемонстрировало, что патогенез DCI включает сигнальный путь NO [18]. Между тем, генетическая изменчивость гена eNOS у человека влияет на риск DCI после SAH, потому что однонуклеотидный полиморфизм T-786C промотора гена eNOS может вызывать более низкую активность eNOS и приводить к большему риску [19, 20].
eNOS также играет важную роль в поддержании CBF после TBI. Пациенты со сниженным уровнем эндогенного eNOS имеют худшие результаты, чем пациенты с аллелями, которые не влияют на уровни eNOS [21]. Предыдущее исследование показало, что иммунореактивность eNOS увеличивается в микроциркуляторном русле, окружающем область воздействия, в первые 3 дня после экспериментальной травмы [22]. Однако у мышей с нокаутом eNOS наблюдается большее снижение CBF, чем у вариантов дикого типа в первые 2 часа после травмы [23].Генетические варианты eNOS также влияют на поддержание CBF после тяжелой ЧМТ.
Ишемический инсульт чаще всего вызывается тромботической или эмболической закупоркой церебральной артерии, которая вызывает прерывание кровотока и гибель тканей. Исследования показали, что тромботические церебральные инфаркты появляются у мышей eNOS +/- уже в возрасте 3–6 месяцев. У мышей с нокаутом eNOS после ишемического инсульта, вызванного окклюзией средней мозговой артерии (MCAO), инфаркты более обширны, чем у вариантов дикого типа [24, 25]. Напротив, введение предшественника NO (L-аргинин) или доноров (нитропруссид натрия (SNP) и 3-морфолиносиднонимин) на модели фокальной ишемии MCAO у крыс улучшило CBF, предотвратило некроз ткани головного мозга и снизило уровни АТФ и глутамата в головном мозге.Это, по-видимому, зависит от времени и ограничивается первыми 30 минутами после начала ишемии. В этот период NO оказывает сосудорасширяющее действие, улучшает CBF и уменьшает размер инфаркта, в то время как он вызывает нервно-сосудистую токсичность за пределами этой точки [25–27]. Более того, eNOS не только способствует расширению сосудов, но также увеличивает пролиферацию и миграцию VSMC, тем самым усиливая артериогенез после инсульта [28]. Активация eNOS обеспечивает защиту от инсульта, сохраняя CBF и подавляя воспаление, агрегацию тромбоцитов, тромбоз и апоптоз [11].Непрерывный произвольный бег обеспечивает долгосрочную активацию eNOS в сосудистой сети и увеличивает количество циркулирующих эндотелиальных клеток-предшественников (EPC) в крови. Тем не менее, это усиливает неоваскуляризацию и CBF за счет eNOS-зависимых механизмов после ишемии головного мозга [29]. eNOS также важен для мобилизации EPC. Нарушение неоваскуляризации у мышей, лишенных eNOS, связано с дефектом мобилизации клеток-предшественников [30].
Сообщалось, что несвязанные eNOS в церебральных артериях участвуют в гипоксически-ишемическом повреждении головного мозга, вызывая окислительный стресс при ишемии [31].Расцепление eNOS происходит из-за отсутствия либо его субстрата L-аргинина, либо кофактора Bh5, который необходим для связывания двух функциональных субъединиц eNOS. Известно, что это обычное явление при многих заболеваниях, таких как гипертония, диабет и гиперхолестеринемия [32, 33]. В несвязанном состоянии поток электронов внутри пептида eNOS перенаправляется на молекулярный кислород, тем самым приводя к неконтролируемому разъединению eNOS и увеличению продукции O 2 • — .Таким образом, нормальная функция eNOS жизненно важна для облегчения цереброваскулярных заболеваний.
Регуляторные механизмы активности eNOS
eNOS имеет гомологичную димеризованную структуру, состоящую из двух идентичных субъединиц. Каждая субъединица содержит С-концевой домен редуктазы и N-концевой домен оксигеназы. Первый содержит сайты связывания для НАДФН, флавинадениндинуклеотида (FAD) и флавинмононуклеотида (FMN), которые находятся в тесной гомологии с редуктазой цитохрома Р-450; последний связывает гем, Bh5, и субстрат L-аргинин.Между этими двумя областями находится связывающий домен кальмодулина (CaM), который играет ключевую роль как в структуре, так и в функции фермента. Для получения полной каталитической активности в физиологических условиях отдельные N-концевые домены оксигеназы и C-концевой редуктазы eNOS должны быть соединены вместе в присутствии гема, Bh5 и комплекса Ca 2+ / CaM [7, 34]. Многие исследования предполагают, что фермент обычно активируется увеличением внутриклеточного Ca 2+ либо за счет притока внеклеточного Ca 2+ , либо за счет высвобождения из мест внутриклеточного хранения.Однако активация eNOS также не зависит от Ca 2+ , хотя активируется механическими силами, включая напряжение сдвига, циклическую деформацию и G-белок [35–37].
Регуляторные механизмы активности eNOS чрезвычайно сложны и могут быть разделены на генетический и белковый (рис. 2). На генетическом уровне экспрессия и стабильность генов eNOS связаны с активностью eNOS. Промотор eNOS содержит большое количество сайтов для связывания факторов транскрипции, включая активаторный белок-1 (AP-1), активаторный белок-2 (AP-2), семейство эндотелина, ядерный фактор-κB (NF-κB), и нейрофибромин 1 (NF-1).Эти комплексы факторов транскрипции могут регулировать экспрессию eNOS. Кроме того, гипоксия, липополисахарид и несколько цитокинов могут индуцировать экспрессию двух видов цитоплазматических белков eNOS. Они связываются с 3′-нетранслируемой областью мРНК eNOS, богатой цитозином, что впоследствии приводит к изменениям конфигурации и активации фермента РНК, что вызывает более низкую стабильность мРНК eNOS и более короткий период полужизни. В настоящее время известно, что многие факторы, такие как гипоксия, эстроген и физические упражнения, влияют на экспрессию eNOS [38-40].На уровне белка регуляторные механизмы в основном включают транслокацию eNOS, образование комплексов и фосфорилирование аминокислотных остатков. Ассоциация eNOS с рецептором брадикинина B2 и кавеолином-1 (Cav-1) в кавеолах и микродоменах эндотелиальной плазматической мембраны приводит к ингибированию активности eNOS. Это ингибирование, по-видимому, является результатом функционального вмешательства в связывание СаМ и перенос электронов [41, 42].
Рис. 2Механизмы регуляции активности eNOS.Механизмы работают как на генетическом, так и на белковом уровне. На генетическом уровне ядерный фактор-κB (NF-κB), белок-активатор-1 (AP-1), белок-активатор-2 (AP-2), Krűppel-подобный фактор 2 (KLF2), семейство эндотелина и Forkhead box O1. (Foxo-1) гены регулируют экспрессию eNOS; на уровне белка несколько факторов, такие как брадикинин, 5-гидрокситриптамин (5-HT), ACh, напряжение сдвига и липопротеин высокой плотности (HDL), способствуют фосфорилированию eNOS с помощью путей PI3K / Akt, AMPK и MAPK. Кроме того, связывание с кальмодулином (CaM) усиливает активацию eNOS, в то время как кавеолин-1 (Cav-1) ингибирует активацию eNOS
Cav-1 на клеточной мембране связывается не только с доменом оксигеназы, но и с доменом редуктазы eNOS.Связывание Cav-1 с доменом редуктазы eNOS ингибирует комбинацию гемового железа с eNOS и предотвращает перенос электронов от редуктазы. В eNOS мотив связывания Cav-1 находится между гемом и доменами связывания CaM, примыкающими к остатку глутамата (Glu-361), необходимому сайту для связывания L-аргинина [42–44]. Таким образом, связывание Cav-1 с eNOS ингибирует доставку и активацию eNOS. Было подтверждено, что два недавно обнаруженных белка, взаимодействующий с eNOS белок (NOSIP) и индуктор трафика eNOS (NOSTRIN), взаимодействуют с eNOS и Cav-1 с образованием тройного комплекса.Затем комплекс индуцирует миграцию eNOS из плазматической мембраны и значительно снижает активность eNOS [45–47].
Настоящие исследования показывают, что фосфорилирование eNOS происходит по остаткам серина, треонина и тирозина. Внутри этих остатков фосфорилирование сайтов Thr495, Ser633 и Ser1177 оказывает наиболее значительное влияние на активность eNOS. Фосфорилирование сайта Thr495 предотвращает связывание CaM с eNOS, тем самым снижая активность eNOS [48], в то время как фосфорилирование сайта Ser1177 увеличивает каталитическую способность eNOS за счет ингибирования разделения CaM и eNOS и увеличивает скорость внутреннего переноса электронов eNOS; фосфорилирование сайта Ser633 также усиливает активность eNOS [49–51].Треониновая протеинкиназа (Akt / PKB) является важной детерминантой фосфорилирования сайта eNOS Ser1177, который участвует в основной активации eNOS и активации его агониста [52, 53].
Akt в основном неактивно располагается в цитоплазме; Транслокация к клеточной мембране вызывает ее активацию и фосфорилирование eNOS [54]. Он напрямую контролируется фосфатидилинозитол-3-киназой серин (PI3K) -зависимым путем фосфорилирования. PI3K привлекает Akt к клеточной мембране и фосфорилирует ее [55].Брадикинин и 5-гидрокситриптамин (5-HT) действуют с мембранными рецепторами, что приводит к увеличению цитоплазматической концентрации Ca 2+ и активации CaM. Сначала он продвигает разъединение eNOS с Cav-1; Затем eNOS соединяется с CaM с образованием комплекса eNOS / CaM и перемещается к цитоплазматической мембране. После транслокации комплекс eNOS / CaM индуцирует фосфорилирование аминокислотных остатков eNOS по сайту Ser1177 и дефосфорилирование по сайту Thr495 через путь Akt / PKB, что приводит к активации eNOS [56-58].
Между тем, после повышения цитоплазматической концентрации Ca 2+ , Ca 2+ / кальмодулин-зависимая протеинкиназа-β (CaMKKβ), альтернативная вышестоящая киназа для аденозин-5′-монофосфат (AMP) -активированной протеинкиназы ( AMPK) и ключевая молекула сигнального пути AMPK, индуцирует фосфорилирование AMPK по сайту Thr172. После активации AMPK способствует фосфорилированию eNOS по сайтам Ser1177 и / или Ser633, усиливает активность eNOS и способствует синтезу NO.Он сдерживается соединением C, специфическим ингибитором AMPK [59–63]. Интересно, что фосфорилирование eNOS Ser1177 выполняет двойную функцию после ишемического инсульта. Экспрессия eNOS, фосфорилирование eNOS Ser1177 и образование мономера eNOS увеличиваются в острой фазе, но фосфорилирование Ser1177 заметно снижается в более поздних фазах ишемии. На этом этапе значительно усиливается разъединение eNOS. Поскольку дисфункция eNOS способствует вторичному повреждению и ингибирует восстановление тканей, дефосфорилирование и образование мономера eNOS могут стать потенциальной терапевтической мишенью церебральной ишемии на более поздних этапах [64].Сайт Ser1177 может быть более важным механизмом нацеливания по сравнению с сайтом Ser633.
Кроме того, eNOS может быть активирован либо напряжением сдвига посредством фосфорилирования Ser1177 через путь PI3K / Akt [50], либо модификацией липопротеинов высокой плотности (HDL) с помощью киназы Akt и митоген-активируемой протеинкиназы (MAPK) [65]. .
Таким образом, сайты Ser1177 и Ser633 могут быть потенциальными терапевтическими сайтами-мишенями. Регулирование фосфорилирования этих двух сайтов и активация eNOS являются регуляторными механизмами CBF.
Кандидаты на таргетную терапию eNOS
Недавние исследования продемонстрировали, что статины обладают потенциалом повышать биодоступность эндотелиевого NO, главным образом за счет повышающей регуляции и активации eNOS. Это опосредуется разнообразной передачей сигналов, такой как Rho / ROCK, PI3K / Akt, кавеолин, ERK 1/2 и так далее [66, 67]. Сходным образом аторвастатин и симвастатин могут увеличивать экспрессию eNOS путем ингибирования Rho-киназы и увеличения Ras-опосредованной активации сигнальных путей PI3K-Akt и ERK1 / 2-RSK, соответственно [68, 69].Кроме того, ингибитор Rho-киназы фасудил улучшает повреждение ткани головного мозга за счет быстрого увеличения фосфорилирования eNOS, подавления дефосфорилирования и мономеризации eNOS в ишемизированном мозге [64].
Набор для анализа активности синтазы оксида азота (колориметрический) (ab211083)
Обзор
Название продукта
Метод обнаружения
Колориметрический
Тип образца
Лизат клеток, лизат ткани
Тип анализа
Количественный
Чувствительность
5 мкЕ
Активность видов
Реагирует с: Млекопитающие, другие виды
Обзор продукции
Набор для анализа активности синтазы оксида азота(колориметрический) (ab211083) обеспечивает точный и удобный метод анализа активности синтазы оксида азота (NOS) в различных образцах.В этом анализе оксид азота (NO), генерируемый NOS, подвергается ряду реакций и реагирует с реагентами Грисса 1 и 2 с образованием окрашенного продукта с сильным поглощением при OD 540 нм. Анализ прост, чувствителен и адаптируется с высокой пропускной способностью и может обнаруживать всего 5 мкЕ активности NOS.
Банкноты
Синтазы оксида азота (EC 1.14.13.39) (NOS) — это семейство ферментов, которые катализируют производство оксида азота (NO) из L-аргинина.Оксид азота (NO) играет важную роль в нейротрансмиссии, регуляции сосудов, иммунном ответе и апоптозе. В присутствии НАДФН, ФАД, ФМН, (6R) -5,6,7,8-тетрагидробиоптерина, кальмодулина и гема NOS катализирует пятиэлектронное окисление гуанидинового азота L-аргинина молекулярным кислородом с образованием NO и L -цитруллин. Существует три изоформы NOS: эндотелиальная (eNOS), нейрональная (nNOS) и индуцибельная (iNOS). nNOS отвечает за производство NO в центральной нервной системе, где NO участвует в клеточной коммуникации и хранении информации.eNOS производит NO в кровеносных сосудах и участвует в регуляции функции сосудов. В отличие от других изоформ, iNOS экспрессируется de novo в условиях окислительного стресса и продуцирует большие количества NO как часть защитного механизма организма.
Платформа
Считыватель микропланшетов
Недвижимость
Инструкции по хранению
Хранить при -80 ° C.См. Протоколы.
Компоненты Идентификатор 100 тестов Аналитический буфер NOS 1 x 25 мл Enhancer (0,5 мкмоль) 1 флакон БУК Кофактор 1 (1 мкмоль) 1 флакон Нитратредуктаза (1 ЕД) 1 флакон Нитритный стандарт (10 мкмоль) 1 флакон Реагент Грисса 1 1 x 10 мл Реагент Грисса 2 1 x 10 мл БДУ (положительный контроль) 1 x 4 мкл NOS Кофактор 2 (25X) 1 x 100 мкл БДУ Буфер для разбавления Красная шапка 1 х 1.5 мл БДУ Подложка 1 x 500 мкл Направления исследований
Функция
Производит оксид азота (NO), который представляет собой молекулу-посредник, выполняющую различные функции по всему телу. В головном мозге и периферической нервной системе NO проявляет многие свойства нейромедиатора.Вероятно, обладает нитрозилазной активностью и опосредует S-нитрозилирование цистеина цитоплазматических белков-мишеней, таких как SRR.
Тканевая специфичность
Изоформа 1 экспрессируется повсеместно: обнаруживается в скелетных мышцах и головном мозге, а также в семенниках, легких и почках, а низкие уровни — в сердце, надпочечниках и сетчатке. В тромбоцитах не обнаруживается. Изоформа 3 экспрессируется только в семенниках. Изоформа 4 обнаруживается в яичках, скелетных мышцах, легких и почках, на низких уровнях в головном мозге, но не в сердце и надпочечниках.
Сходства последовательностей
Принадлежит к семейству NOS.
Содержит 1 FAD-связывающий домен FR-типа.
Содержит 1 флаводоксиноподобный домен.
Содержит 1 домен PDZ (DHR).Домен
Домен PDZ в N-концевой части изоформы нейронов участвует во взаимодействии белок-белок и отвечает за нацеливание nNo на синаптические мембраны в мышцах.Опосредует взаимодействие с VAC14.
Пост-трансляционные
модификацииубиквитинированный; опосредовано STUB1 / CHIP в присутствии Hsp70 и Hsp40 (in vitro).
Сотовая локализация
Клеточная мембрана> сарколемма. Клеточная проекция> дендритный шип. В скелетных мышцах он локализуется под сарколеммой быстро сокращающихся мышечных волокон, связываясь с гликопротеиновым комплексом дистрофина.В нейронах, обогащенных дендритными шипами.
- Информация от UniProt
Альтернативные названия
- 2310005C01Rik
- BNOS
- Учредительный NOS
- EC 1.14.13.39
- IHPS 1
- IHPS1
- N-NOS
- NC-NOS
- нейрональная синтаза оксида азота
- Нейронная БДУ
- Синтаза оксида азота, нейрональная, в том числе
- Синтаза оксида азота 1
- Синтаза оксида азота 1 (нейрональная)
- Синтаза оксида азота, мозг
- Синтаза оксида азота, нейроны полового члена, в том числе
- NNOS
- НЕТ
- НОМЕР
- № 1
- БДУ тип I
- NOS-I
- NOS1
- NOS1_HUMAN
- Пептидилцистеин-S-нитрозилаза NOS1
посмотреть все
Изображения
Набор для анализа активности синтазы оксида азота (колориметрический) (ab211083)
Набор для анализа активности синтазы оксида азота (колориметрический) (ab211083).Типичная стандартная калибровочная кривая.
Набор для анализа активности синтазы оксида азота (колориметрический) (ab211083)
Набор для анализа активности синтазы оксида азота (колориметрический) (ab211083). Измерение активности положительного контроля NOS (10 мкл) по сравнению с бланком (буфер для анализа).
Набор для анализа активности синтазы оксида азота (колориметрический) (ab211083)
Набор для анализа активности синтазы оксида азота (колориметрический) (ab211083).Обнаружение эндогенной активности NOS в лизатах клеток J774.1A. Клетки J774 стимулировали 200 нг / мл LPS и 100 нг / мл мышиного IFN-гамма. Включен нестимулированный контроль. Готовили клеточные лизаты (225 мкг) и анализировали в соответствии с протоколом набора
.
Листы данных и документы
SDS скачать
Страна / регион Выберите страну / регион
Язык Выбор языка
Скачать брошюру
Список литературы (7)
ab211083 упоминается в 7 публикациях.
- Shaldoum F et al. Иммуномодулирующие эффекты пчелиной пыльцы на индуцированную доксорубицином иммуносупрессию костного мозга / селезенки у крыс. J Food Biochem 45: e13747 (2021). PubMed: 33949702
- Wang J & Liu G Защитный эффект микроРНК-340-5p против кислородно-глюкозной депривации / реперфузии в клетках PC12 посредством нацеливания на дифференцировку нейронов 4. Mol Med Rep 22: 964-974 (2020). PubMed: 32468054
- Картикеян В и др. Оценка белков ARG2 и NOS1 человека, связанных с варикоцеле, и вычислительный анализ влияния его nsSNP. Int J Biol Macromol 164: 735-747 (2020). PubMed: 32693129
- Szabó R et al. Потенциальное участие анандамида в модуляции систем HO / NOS: женщины, менопауза и «медицинские каннабиноиды». Int J Mol Sci 21: Н / Д (2020). PubMed: 33233803
- Hanna A et al. Ингибирование передачи сигналов Hedgehog перепрограммирует дисфункциональную иммунную микросреду при раке груди. Онкоиммунология 8: 1548241 (2019). PubMed: 30723576
- Fu Y et al. Гастропротекторные и противоязвенные эффекты оксиматрина на нескольких моделях язвы желудка у крыс: возможные роли антиоксидантных, противовоспалительных механизмов и механизмов выживания. Phytother Res Н / Д: Н / Д (2018). PubMed: 30024074
- Веремейко Т и др. Ген-2 ответа раннего роста необходим для активации и пластичности макрофагов M1 и M2 путем модуляции фактора транскрипции CEBPß. Front Immunol 9: 2515 (2018). PubMed: 30443252