Хронический лейкоз у взрослых
Лейкоз (лейкемия) является злокачественным заболеванием лейкоцитов (белых кровяных телец). Он возникает в костном мозге и затем поражает кровь, лимфатические узлы, селезенку, печень, центральную нервную систему (ЦНС) и другие органы. Лейкоз может развиваться как у взрослых, так и детей.
Лейкоз подразделяется на многие типы и подтипы, которые имеют свои особенности клинического течения, лечения и прогноза (исхода).
Для лучшего понимания различных типов лейкоза необходимо иметь основные представления о кровеносной и лимфатической системах.
Костный мозг расположен внутри костей. В нем образуются различные клетки крови.
Ранние (молодые, или незрелые) клетки крови носят название стволовых. Эти клетки созревают и превращаются в эритроциты (красные кровяные тельца), лейкоциты и тромбоциты.
Эритроциты переносят кислород из легких в другие ткани и организма. Они же выводят углекислый газ – продукт жизнедеятельности клеток.
Лейкоциты помогают в борьбе против вирусов и бактерий. Различают несколько типов и подтипов лейкоцитов, каждый из которых играет особую роль в защите против инфекций.
Выделяют три основных типа лейкоцитов: гранулоциты, моноциты и лимфоциты.
Тромбоциты предотвращают кровотечения при порезах и ушибах.
Лимфатическая система включает в себя лимфатические сосуды, лимфатические узлы и лимфу.
Лимфатические сосуды напоминают вены, но переносят не кровь, а лимфу – прозрачную жидкость, содержащую тканевую жидкость, продукты жизнедеятельности организма и клетки иммунной системы.
Лимфатические узлы располагаются вдоль лимфатических сосудов и накапливают в себе клетки иммунной системы. При воспалении и некоторых других заболеваниях они могут увеличиваться в размерах.
Типы лейкозов
Различают:
острый и хронический лейкозы
лимфоцитарный (лимфобластный) и миелоидный лейкозы
При остром лейкозе болезнь быстро прогрессирует, и хотя клетки растут быстро, они не способны к созреванию.
При хроническом лейкозе опухолевые клетки напоминают нормальные, но отличаются от них. Они живут слишком долго и мешают образованию некоторых видов лейкоцитов.
Лимфоцитарный и миелоидный лейкозы получили свое название в соответствии с клетками, из которых они возникли.
| Острый лимфоцитарный лейкоз (ОЛЛ) (лимфобластный) | Острый миелоидный лейкоз (ОМЛ) (нелимфоцитарный, ОНЛЛ) |
| Встречается чаще у детей, чем у взрослых и составляет более 50% лейкозов детей. | Возникает у детей и взрослых, составляя менее половины всех случаев лейкозов детского возраста. |
| Хронический лимфоцитарный лейкоз (ХЛЛ) | Хронический миелоидный лейкоз (ХМЛ) |
| Поражает взрослых и встречается в 2 раза чаще ХМЛ. | Выявляется преимущественно у взрослых и очень редко у детей и встречается в 2 раза реже ХЛЛ. |
Как часто встречается хронический лейкоз у взрослых?
Хронический лимфоцитарный лейкоз (лимфолейкоз) – наиболее распространенный вид лейкоза в странах Европы и Северной Америки. На его долю приходится 30% среди всех лейкозов.
На его долю приходится 30% среди всех лейкозов.
Ежегодная заболеваемость ХЛЛ в этих странах составляет 3-3,5 на 100 тыс. населения, а среди лиц старше 65 лет – до 20 на 100 тыс.населения.
Около 70% пациентов заболевают между 50 и 70 годами. Средний возраст к началу заболевания составляет 55 лет. Только менее 10% заболевают в возрасте моложе 40 лет.
Мужчины болеют в 2 раза чаще женщин.
Хронический миелоидный лейкоз составляет около 20% среди всех лейкозов. В странах Европы и Северной Америки по частоте распространения ХМЛ занимает 3 место после острых лейкозов и ХЛЛ. Ежегодная заболеваемость составляет 1-1,5 на 100 тыс. населения во всех странах и остается практически стабильной на протяжении последних 50 лет.
Мужчины заболевают несколько чаще женщин, составляя 55-60% больных. Половина пациентов заболевают в возрасте 30-50 лет, чаще всего между 30-40 годами. У детей типичный ХМЛ встречается редко, составляя не более 1-2% случаев детских лейкозов.
Причины возникновения хронического лейкоза и возможность его предотвращения
В настоящее время известны некоторые факторы риска, связанные с развитиемразвитием хронического лейкоза. Так, воздействие высоких доз радиации при взрыве атомной бомбы или аварии на атомном реакторе повышает риск хронического миелоидного лейкоза, но не хронического лимфоцитарного лейкоза.
Длительный контакт с гербицидами или пестицидами среди сельских жителей может повысить риск возникновения хронического лимфоцитарного лейкоза.
Высоковольтные линии передач, возможно, являются фактором риска развития лейкоза.
У большинства больных лейкозом не выявлены факторы риска, поэтому не существует способов предотвращения этого заболевания. Исключение составляет курение, которое повышает риск возникновения лейкоза.
Диагностика хронического лейкоза
В настоящее время еще не разработаны методы раннего выявления хронического лейкоза. При появлении у необычных симптомов необходимо срочно обратиться к врачу.
У 50% больных хроническим лейкозом отсутствуют какие-либо симптомы в момент выявления заболевания. У этих пациентов заболевание диагностируется по данным анализа крови, выполненного по другому поводу.
Общие симптомы хронического лейкоза могут включать повышенную утомляемость, слабость, потерю веса, повышение температуры и боли в костях. Большинство из этих симптомов связаны со снижением количества клклеток крови.
Анемия (малокровие) возникает в результате уменьшение количества эритроцитов, что приводит к одышке, повышенной утомляемости и бледности кожи.
Снижение числа нормальных лейкоцитов повышает риск инфекционных заболеваний. У больных лейкозом количество лейкоцитов может быть значительно повышено, однако эти опухолевые клетки не защищают от инфекции.
Уменьшение числа тромбоцитов сопровождается кровоизлияниями, кровотечениями из носа и десен.
Распространение лейкоза из костного мозга в другие органы и центральную нервную систему может привести к головной боли, слабости, судорогам, рвоте, нарушению зрения.
Лейкоз может сопровождаться увеличением лимфатических узлов, печени и селезенки.
Методы диагностики
Анализ крови. По количеству клеток крови и их виду под микроскопом можно заподозрить лейкоз. У большинства больных хроническим лейкозом имеется повышенное количество лейкоцитов, снижение числа эритроцитов и тромбоцитов.
Биохимический анализ крови помогает уточнить функцию почек и состав крови.
Исследование костного мозга дает возможность установить диагноз лейкоза и оценить эффективность лечения.
Спинно-мозговая пункция позволяет выявить опухолевые клетки в спинно-мозговой жидкости и провести лечение путем введения химиопрепаратов.
С целью уточнения типа лейкоза используются специальные методы исследования: цитохимия, проточная цитометрия, иммуноцитохимия, цитогенетика и молекулярно-генетическое исследование.
Рентгенологические исследования грудной клетки и костей позволяют выявить поражение лимфатических узлов средостения, костей и суставов.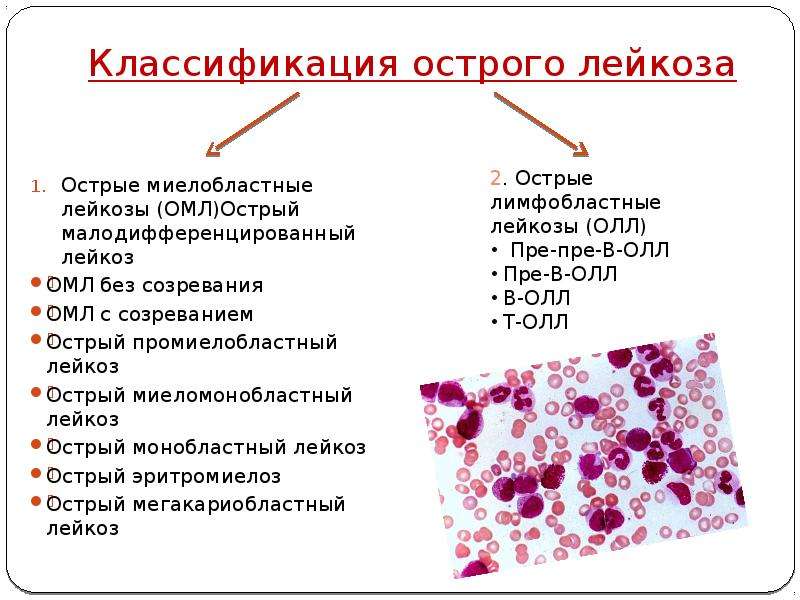
Компьютерная томография (КТ) дает возможность обнаружить поражение лимфатических узлов в грудной полости и животе.
Магнитно-резонансная томография (МРТ) особенно показана при исследовании головного и спинного мозга.
Ультразвуковое исследование (УЗИ) позволяет отличить опухолевые и кистозные образования, выявить поражение почек, печени и селезенки, лимфатических узлов.
Стадии хронического лейкоза
В зависимости от степени распространения заболевания при большинстве злокачественных опухолей определяется стадия – от 1 до 4.
Однако, лейкоз является системным заболеванием, при котором к моменту диагностики имеется поражение костного мозга и других органов, поэтому при лейкозе стадия не определяется.
Для оценки прогноза (исхода) заболевания учитываются другие характеристики, влияющие на выбор тактики лечения.
Лечение хронического лейкоза
Лечение больных хроническим лейкозом зависит от типа заболевания и прогностических факторов.
Лекарственный метод является основным при лечении хронического лейкоза.
Выбор тактики лечения больных ХЛЛ зависит как от распространенности опухолевого процесса, так и от наличия определенных симптомов. С учетом этих факторов, а также клеточных и хромосомных изменений, больные подразделяются на группы риска.
Группа низкого риска.
Прогноз (исход) заболевания у пациентов этой группы благоприятный. Средняя выживаемость составляет 20-25 лет. Обычно лечение не назначается, а рекомендуется тщательное наблюдение. Лишь в случае дальнейшего развития болезни или появления неприятных симптомов применяется лечение.
Группа промежуточного и высокого риска.
У больных при отсутствии симптомов от лечения можно временно воздержаться. При появлении признаков прогрессирования заболевания или новых симптомов может быть назначена терапия.
Химиоптерапия проводится, как правило, противоопухолевым препаратом хлорамбуцилом. При появлении выраженных побочных эффектов этот препарат может быть заменен на циклофосфамид. Иногда используются стероидные препараты (преднизон).
Иногда используются стероидные препараты (преднизон).
У некоторых больных применяют комбинированную химиотерапию с включением циклофосфамида, доксорубицина, винкристина.
Флюдарабин применяется обычно при рецидиве (возврате) заболевания после лечения комбинацией препаратов. У молодых больных этот препарат можно применять в самом начале лечения. При увеличении селезенки или лимфатических узлов возможно назначение лучевой терапии в низких дозах. В случае появления выраженных симптомов, связанных со значительным увеличением селезенки, выполняется удаление селезенки.
Больным с большим количеством лейкоцитов, нарушающим кровоток, до химиотерапии показан лейкаферез (удаление избытка лейкоцитов, включая опухолевые клетки). Эффект наступает быстро, но бывает временным.
В редких случаях применяется трансплантация стволовых клеток, однако эффективность данного метода еще не доказана.
Иногда ХЛЛ может трансформироваться (превращаться) в острый лейкоз или агрессивную неходжкинскую лимфому (лимфосаркому).
Лечение хронического миелоидного лейкоза (миелолейкоза) в зависимости от фазы заболевания.
Выбор тактики лечения больных с ХМЛ зависит от фазы заболевания (хроническая, акселерации, бластный криз), возраста больного, прогностических факторов и наличия подходящего донора.
Хроническая фаза.
Применение препарата гливек (иматиниб) приводит к достижению полного эффекта у 90% больных ХМЛ.
До этого использовалась химиотерапия высокими дозами препаратов в сочетании с тотальным облучением и трансплантацией стволовых клеток.
Фаза обострения.
Применение гливека может привести к достижению ремиссии (отсутствию признаков болезни), однако период улучшения длится недолго. Использование интерферона также не позволяет получать длительные ремиссии. У 20% больных отмечается положительный ответ на химиотерапию, но он длится не более 6 месяцев.
Приблизительно 15% больных в этой фазе ХМЛ живут в течение нескольких лет после трансплантации стволовых клеток.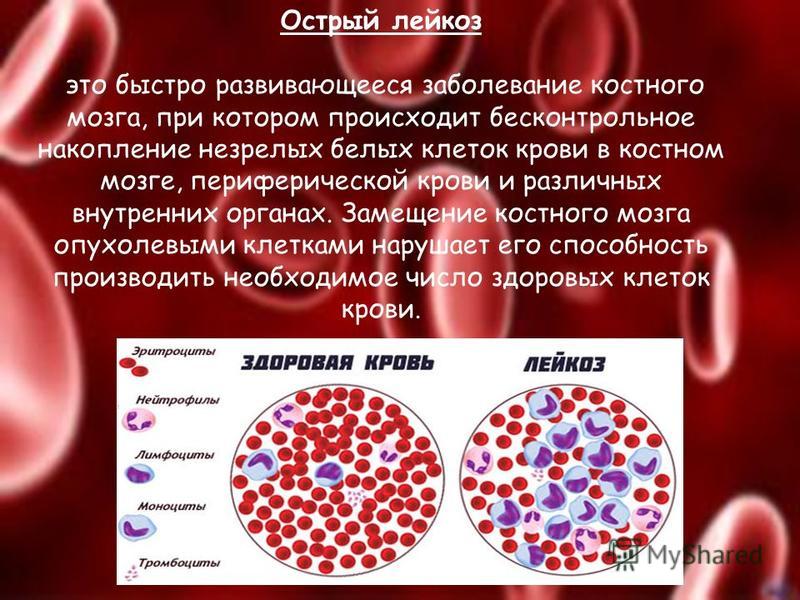 Эту процедуру лучше выполнять молодым больным после эффективной химиотерапии.
Эту процедуру лучше выполнять молодым больным после эффективной химиотерапии.
Бластный криз.
В этой фазе болезни опухолевые клетки напоминают таковые при остром миелоидном лейкозе (ОМЛ), причем они мало чувствительны к химиотерапии. Если же и достигается положительный эффект, то он бывает кратковременным. В этом случае возможно применение трансплантации стволовых клеток.
У некоторых больных опухолевые клетки напоминают клетки острого лимфобластного лейкоза (ОЛЛ), которые более чувствительны к химиотерапии. Поэтому использование винкристина, доксорубицина и преднизона может привести к ремиссии.
При поражении центральной нервной системы у больных ХМЛ применяется цитарабин, вводимый в спинномозговой канал, или облучение головного мозга.
заболевание, симптомы, лечение, причины, диагностика
Лейкоз — клональное злокачественное заболевание кроветворной системы. Встречаются также названия — лейкемия, белокровие, иногда «рак крови». К лейкозам относится обширная группа заболеваний, различных по своему происхождению. Опухоли при лейкозе возникают из кроветворных клеток и поражают костный мозг.
К лейкозам относится обширная группа заболеваний, различных по своему происхождению. Опухоли при лейкозе возникают из кроветворных клеток и поражают костный мозг.
По характеру течения лейкозы различают как острые, из незрелых клеток (бластов), и хронические, из созревающих и зрелых клеток.
Острые лейкозы по цитогенезу подразделяются на:
лимфобластный,
миелобластный,
монобластный,
миеломонобластный,
эритромиелобластный,
мегакариобластный,
недифференцированный.
Хронические лейкозы представлены лейкозами:
Для всех острых лейкозов характерны нарастающая «беспричинная» слабость, недомогание, иногда одышка, головокружение, обусловленные анемией. Повышение температуры тела, интоксикация — частые симптомы нелимфобластных острых лейкозов. Увеличение лимфатических узлов, печени и селезенки в развернутой стадии встречается не при всех острых лейкозах.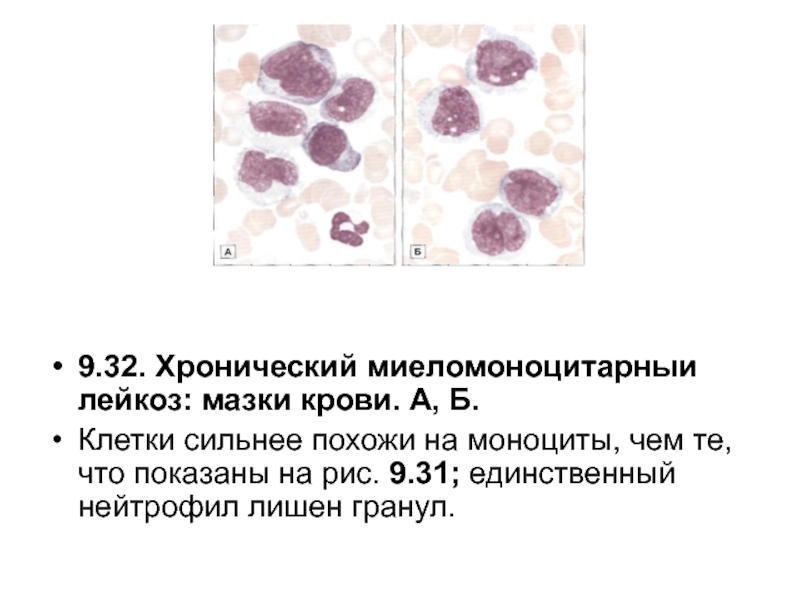 Нередок геморрагический синдром, обусловленный прежде всего тром-боцитопенией.
Нередок геморрагический синдром, обусловленный прежде всего тром-боцитопенией.
При хроническом миелолейкозе опухолевым процессом поражается как гранулоцитарный, так и тромбоцитарный и эритроцитарный ростки костного мозга. Родоначальница опухоли -клетка-предшественница миелопоэза. Процесс может распространяться на печень, селезенку, а в терминальной стадии пораженной может оказаться любая ткань. В начале развернутой стадии у больного отсутствуют жалобы, не увеличена или незначительно увеличена селезенка, состав периферической крови изменен. Развернутая стадия может продолжаться в среднем 4 года. При правильной терапии состояние больных остается удовлетворительным, они сохраняют трудоспособность, ведут обычный образ жизни при амбулаторном наблюдении и лечении. В терминальной стадии течение хронического миелолейкоза приобретает черты злокачественности.
Хронический лимфолейкоз представляет собой доброкачественную опухоль иммунокомпетентной системы.
Хронический моноритарный лейкоз относится к редким формам лейкозов.
Любые формы лейкозов являются очень серьезными и опасными для жизни заболеваниями, поэтому необходимо их вовремя диагностировать.
Преимущества услуги
Удобный график работы
Работаем до позднего вечера, чтобы вам было удобно заняться своим здоровьем после работы
Отсутствие очередей
Система записи пациентов отлажена за много лет работы и действует так, что вас примут точно в выбранное время
Уютный интерьер
Нам важно, чтобы пациенты чувствовали себя комфортно в стенах клиники, и мы сделали все, чтобы окружить вас уютом
Внимание к пациенту
К вашим услугам – внимательный персонал, который ответит на любой вопрос и поможет сориентироваться
ХРОНИЧЕСКИЙ МИЕЛОЛЕЙКОЗ
Новые препараты при хроническом миелолейкозе
Santos FP, Quintás-Cardama A
Терапия первой линии хронического миелолейкоза дазатинибом, нилотинибом или иматинибом
Wei G, Rafiyath S, Liu D
CD34+ клетки уменьшили эффективность иматиниба из-за низкой активности OCT-1 при хроническом миелобластном лейкозе
Engler JR, Frede A, Saunders VA, Zannettino AC, Hughes TP, White DL
Терапия дазатинибом при хронической фазе хронического миелоидного лейкоза: Анализ ответов согласно существующим ранее мутациям BCR-ABL
Muller MC, Cortes JE, Kim D-W, Druker BJ, Erben P, Pasquini R, Branford S, Hughes TP, Radich JP, Ploughman L, Mukhopadhyay J, Hochhaus A
Динамика и управление цитопенией, связанной с терапией дазатинибом, у пациентов с хроническим миелоидным лейкозом в хронической фазе после неудачи иматиниба
Quintás-Cardama A, De Souza Santos FP, Kantarjian H, O’Brien S, Faderl S, Awais A, Borthakur G, Cortes J
Значение субоптимального ответа на иматиниб для отдаленного результата у пациентов с хроническим миелоидным лейкозом в хронической фазе
Alvarado Y, Kantarjian H, O’Brien S, Faderl S, Borthakur G, Burger J, Wierda W, Garcia-Manero G, Shan J, Cortes J
Стратегия использования офатумумаба в первой линии терапии хронического лимфоцитарного лейкоза
Zoler ML
Предсказатели первичной резистентности к иматинибу при хроническом миелоидном лейкозе отличны от факторов при вторичной резистентности
Zhang WW, Cortes JE, Yao H, Zhang L, Reddy NG, Jabbour E, Kantarjian HM, Jones D
Специфический кариотип микроРНК при хроническом лимфоцитарном лейкозе
Visone R, Rassenti LZ, Veronese A, Taccioli C, Costinean S, Aguda BD, Volinia S, Ferracin M, Palatini J, Balatti V, Alder H, Negrini M, Kipps TJ, Croce CM
Отсроченное достижение цитогенетического и молекулярного ответа у пациентов в ранней хронической фазе хронического миелоидного лейкоза, получающих высокие или стандартные дозы иматиниба, связано с увеличенным риском прогрессирования
Quintas-Cardama A, Kantarjian H, Jones D, Shan J, Borthakur G, Thomas D, Kornblau S, O’Brien S, Cortes J
Идентификация и причины неудач лечения иматинибом при хроническом миелоидном лейкозе: практический обзор клинических рекомендаций и доступных препаратов
SL Goldberg, A Masood
Терапия первой линии при хроническом миелоидном лейкозе: прошлое, настоящее и будущее
C Pavlovsky, H Kantarjian, JE Cortes
Хронический миелолейкоз: принять решение. Вклад таргетной терапии в лечение ХМЛ
Вклад таргетной терапии в лечение ХМЛ
M Copeman
Выбирая терапию второй линии
G Saglio
Пример успешного лечения нилотинибом пациента в ХФ ХМЛ с резистентностью к α-интерферону и иматинибу
Г.А. Гусарова
Эффективность терапии нилотинибом у пациентки с резистентным к иматинибу Ph+ ХМЛ в хронической фазе
Э.Г. Ломаиа
Нилотиниб при фазе акселерации ХМЛ
G Rosti
Нилотиниб при хронической фазе ХМЛ
M Copeman
ХМЛ: оптимизация дозы иматиниба
А.Ю. Зарицкий
Мониторинг и интерпретация результатов молекулярного ответа при ХМЛ
A.Г. Туркина
Нилотиниб – новый этап успеха в терапии хронического миелолейкоза
Э.Г. Ломаия, А.Ю. Зарицкий
Случай успешной терапии нилотинибом пациентки с Ph-позитивным хроническим миелолейкозом в хронической фазе с резистентностью к интерферону и Гливеку
Н.А. Афанасьева, Г.А. Гусарова
Расширенный доступ к нилотинибу в рамках клинического исследования (ENACT) у взрослых пациентов с хроническим миелоидным лейкозом в фазе бластного криза, акселерации или хронической фазе с резистентностью к иматинибу или непереносимостью иматиниба: предварительный анализ безопасности
F Nicolini, G Alimena, Haifa-Kathrin Al-Ali, G Smith, Maria Luisa Veronese
Нилотиниб имеет минимальную перекрестную непереносимость с иматинибом у пациентов с Ph+-хроническим миелоидным лейкозом в хронической фазе или фазе акселерации
J Cortes, E Jabbour, A Hochhaus, Ph. le Coutre, M Baccarani, KN Bhalla, G Ossenkoppele, N Gattermann, A Haque, N Gallagher, F Giles, HM Kantarjian
Высокая эффективность и безопасность Нилотиниба у пациентов в хронической фазе хронического миелоидного лейкоза с резистентностью к иматинибу или непереносимостью иматиниба
HM Kantarjian, A Hochhaus, J Cortes, G Martinelli, Ph. le Coutre, KN Bhalla, FJ Giles, G Ossenkoppele, N Gattermann, A Haque, N Gallagher, M Baccarani
Шестилетние результаты исследования IRIS: длительная выживаемость и снижение частоты трансформаций у первично диагностированных пациентов с хроническим миелоидным лейкозом в хронической фазе, получающих лечение иматинибом
A Hochhaus, BJ Druker, RA Larson, SG O’Brien, I Gathmann, F Guilhot
Организация терапии хронического миелолейкоза. Первый общероссийский регистр больных хроническим миелолейкозом: анализ и перспективы
О.Ю. Виноградова, А.Г. Туркина, Н.Д. Хорошко
Рекомендации по ведению пациентов с хроническим миелоидным лейкозом (ХМЛ): Рекомендации European LeukemiaNet (ELN)
Рекомендации по ведению больных с хроническим миелолейкозом (ХМЛ)
Bacсarani и соавторы, Европейское Общество по Лечению Лейкозов (ELN)
Рекомендации по лечению больных хроническим миелолейкозом (ХМЛ) с резистентностью к иматинибу или его непереносимостью с помощью ингибитора киназы препарата Спрайсел (дазатиниб)
Дж.Х. Липтон, Д.Л. Форрест, С. Гамбакорти-Пассэрини, П. Лоневиль, Б. Лебер, Р. Тернер
Новые возможности в терапии хронического миелолейкоза: дазатиниб
М.А. Волкова
Dasatinib or high-dose imatinib for chronic-phase chronic myeloid leukemia after failure of first-line imatinib: a randomized phase 2 trial
Kantarjian H, Pasquini R, Hamerschlak N, Rousselot P, Holowiecki J, Jootar S, Robak T, Khoroshko N, Masszi T, Skotnicki A, Hellmann A, Zaritsky A, Golenkov A, Radic Jh, Hughes T, Countouriotis A, Shah N
Сравнение дазатиниба и высоких доз иматиниба в лечении хронической фазы хронического миелолейкоза при неэффективности стандартных доз иматиниба: рандомизированное исследование фазы II
Х. Кантарджян, Р. Паскуини, Н. Хамершлак, Ф. Руссело, Е. Холовецки, С. Джутар, Т. Робак, Н. Хорошко, Т. Массзи, А. Скотницки, А. Хеллманн, А. Зарицкий, А. Голенков, Дж. Радич, Т. Хьюз, А. Кунтуриотис, Н. Шах
Результаты использования высоких доз иматиниба мезилата при промежуточном риске SOKAL у пациентов с хроническим миелоидным лейкозом в ранней хронической фазе: II фаза исследования GIMEMA CML WP
Castagnetti F, Palandri F, Amabile M, Testoni N, Luatti S, Soverini S, Iacobucci I, Breccia M, Rege Cambrin G, Stagno F, Specchia G, Galieni P, Iuliano F, Pane F, Saglio G, Alimena G, Martinelli G, Baccarani M, Rosti G
Увеличение дозы иматиниба мезилата приводит к длительным ремиссиям у пациентов с цитогенетическими рецидивами, ранее получавших стандартную дозу иматиниба
Jabbour E, Kantarjian HM, Jones D, Shan J, O’Brien S, Reddy N, Wierda WG, Faderl S, Garcia-Manero G, Verstovsek S, Rios MB, Cortes J
Беременность близнецами у пациентки с ХМЛ на фоне терапии Иматинибом
Meera V, Jijina F, Shrikande M, Madkaikar M, Ghosh K
Хронический миелолейкоз: клинические рекомендации ESMO по диагностике, лечению и наблюдению
Hochhaus A, Dreyling M, on behalf of ESMO Guidelines Working Group
Хронические миелолейкозы | Справочник лекарственных препаратов Компендиум
Заболевания, относящиеся к группе хронических миелопролиферативных заболеваний (ХМПЗ), возникают в результате злокачественной трансформации полипотентной гемопоэтической стволовой клетки костного мозга и последующей клональной пролиферации клеток одной или более линий миелопоэза, сохраняющих способность к дифференцировке [1–3]. Различные формы ХМПЗ имеют ряд сходных и перекрывающихся морфологических и клинико-гематологических признаков (спленомегалия, лейкоцитоз, тромбоцитоз, увеличение количества мегакариоцитов и развитие фиброза в костном мозге). Возникновение фиброза, а в ряде случаев и явлений склероза, носит реактивный характер. Обусловлено оно пролиферацией фибробластов, являющихся основными клеточными элементами кроветворного микроокружения костного мозга, не относящихся к клону злокачественных клеток.
В то же время имеются и существенные различия клинико-лабораторных данных, на которых основывается современная классификация ХМПЗ [4–6]. При изучении мазков периферической крови и костного мозга, гистологическом исследовании трепанобиоптатов, применении цитогенетических и молекулярно-биологических методов удается выделить следующие основные формы ХМПЗ (табл. 21).
Хронический миелолейкоз
Хронический миелолейкоз (ХМЛ) является клональным злокачественным процессом. У 95% больных в клетках костного мозга и периферической крови определяется так называемая филадельфийская (Ph’) хромосома, возникающая в результате транслокации генетического материала между хромосомами 9 и 22 — t (9; 22) (q34.1; q11.21). У 5% больных с ХМЛ Ph’ хромосома не определяется (так называемые Ph’ негативные случаи). Но при этом у некоторых больных выявляются характерные аномалии BCR-ABL [8].
На долю ХМЛ приходится около 15–20% всех случаев лейкозов у взрослых и 5% у детей. В США ежегодно регистрируется 3400 новых случаев заболевания ХМЛ [2]. Возрастной пик заболеваемости приходится на 4–5-е десятилетие жизни. Среди больных несколько преобладают лица мужского пола.
В гематологическом плане заболевание характеризуется выраженным лейкоцитозом, сочетающимся с базофилией и эозинофилией. Лейкоцитоз обусловлен увеличением в периферической крови количества зрелых и незрелых нейтрофилов [2, 3, 6].
Хроническая фаза заболевания
Общее количество лейкоцитов в периферической крови колеблется в широких пределах, но обычно выше 50•109/л. У 70–90% больных количество лейкоцитов превышает 100•109/л, а у 25% — выше 350•109/л [1–4, 6]. Выраженный лейкоцитоз часто наблюдается при ХМЛ у детей. В хронической фазе заболевания содержание миелобластов обычно колеблется в пределах 2–3%, а общее количество промиелоцитов и миелобластов не превышает 15–20% от общего количества лейкоцитов в периферической крови или костном мозге. Количество незрелых клеточных элементов гранулоцитарного ряда (промиелоцитов и миелоцитов) увеличивается по мере прогрессирования процесса при одновременном уменьшении процентного содержания палочкоядерных и сегментоядерных лейкоцитов. В нейтрофилах периферической крови и костного мозга в хронической фазе ХМЛ, как правило, не наблюдается диспластических изменений. У некоторых больных отмечаются выраженная эозинофилия и базофилия (так называемая эозинофило-базофильная ассоциация) и наличие многочисленных незрелых клеток эозинофильного и базофильного ряда. У большинства больных с ХМЛ определяется также абсолютный моноцитоз. Выраженный моноцитоз в ранней фазе заболевания позволяет предположить наличие хронического миеломоноцитарного лейкоза (ХММЛ). Во время установления диагноза у многих больных с ХМЛ определяется нормоцитарная нормохромная анемия, признаки анизоцитоза и пойкилоцитоза. Анемия прогрессирует по мере увеличения количества лейкоцитов в периферической крови. Одновременно в крови определяется небольшое количество ядросодержащих клеточных элементов эритробластического ряда. Почти у 50% больных с ХМЛ отмечается тромбоцитоз. Количество тромбоцитов в крови увеличивается по мере развития заболевания, нередки случаи, когда тромбоцитоз составляет 1000•109/л. В мазках периферической крови больных обнаруживаются гигантские формы тромбоцитов, пластинки с аномальной грануляцией, ядра мегакариоцитов.
В периферической крови больных нередко еще за несколько месяцев до манифестации ХМЛ определяются псевдопельгеровские лейкоциты. Помимо ХМЛ, эта приобретенная аномалия гранулоцитов может обнаруживаться у больных с идиопатическим миелофиброзом, при ОМЛ, иногда при неходжкинских злокачественных лимфомах, инфекционных процессах, действии ряда токсических веществ.
В костном мозге больных с ХМЛ обнаруживается гиперклеточность, обусловленная преобладанием нейтрофилов и незрелых клеток гранулоцитарного ряда. Резко увеличено соотношение клеток гранулоцитарного и эритробластического ряда, составляющее нередко 20:1 вместо 3:1 в норме. В костном мозге определяется тот же спектр клеток гранулоцитарного ряда, что и в периферической крови, но с бо’льшим преобладанием незрелых клеток (промиелоцитов и миелоцитов). Отмечается преимущественно нормобластический эритропоэз, но у отдельных больных в хронической фазе заболевания — мегалобластический и признаки дисэритропоэза. Количество мегакариоцитов увеличено, как и при многих других миелопролиферативных заболеваниях. Палочки Ауэра в цитоплазме клеток, в отличие от ОМЛ, определяются редко. Их появление может служить предвестником развития бластного криза.
Приблизительно у 30% больных ХМЛ в костном мозге и селезенке определяются крупные клетки с пенистой цитоплазмой и эксцентрично расположенным ядром, напоминающие клетки, обнаруживаемые при болезни Гоше. Эти псевдо-Гоше-клетки изредка встречаются также при врожденной дисэритропоэтической анемии, множественной миеломе, иммунобластной лимфоме, лимфогранулематозе. Считается, что их появление обусловлено неспособностью клеточных гликоцереброзидаз расщеплять гликоцереброзиды, образующиеся в больших количествах при усилении распада лейкоцитов. Подобно истинным клеткам Гоше они PAS-положительны, окрашиваются масляным красным О, СЧВ, обладают активностью КФ [2, 3, 5].
В костном мозге больных ХМЛ при окраске по Романовскому—Гимзе встречаются также крупные, так называемые цвета голубого моря гистиоциты. Эти клетки с яркой сине-зеленой цитоплазмой, как и псевдо-Гоше-клетки, дающие положительную реакцию на гликоген и липиды, изредка выявляются при идиопатической тромбоцитопенической пурпуре, ОМЛ, ОЛЛ, неходжкинских злокачественных лимфомах. Возможно, что оба типа клеток в действительности представляют две разные стадии развития макрофагов, содержащих остатки фагоцитированных нейтрофилов [1].
Получение и исследование пунктатов костного мозга, как и периферической крови, не только обязательно для установления диагноза ХМЛ. Оно позволяет провести цитогенетические исследования, определить реарранжировку генов BCR и ABL, изучить характер роста клеток-предшественников в культуре in vitro [6–8].
По данным гистологического изучения трепанобиоптатов костный мозг представляется выраженно гиперклеточным за счет увеличения количества зрелых и незрелых клеток гранулоцитарного ряда и мегакариоцитов. Наибольшее количество промиелоцитов и миелоцитов располагается вблизи эндоста и периваскулярно. Мегакариоциты распределяются в срезах равномерно или образуют кластеры. По мере прогрессирования заболевания усиливается пролиферация клеток мегакариоцитарного ряда и могут выявляться многочисленные микромегакариоциты. Миелофиброз, выраженность которого усиливается по мере развития процесса, может определяться и на ранних стадиях заболевания. Увеличивается количество ретикулиновых волокон, располагающихся в виде отдельных фокусов преимущественно в периваскулярных пространствах или диффузно. Выраженность фиброза в начальной стадии развития ХМЛ коррелирует со спленомегалией, уровнем гемоглобина, процентным содержанием бластов в костном мозге и периферической крови, дополнительными кариотипическими аномалиями. Фиброз, обусловленный наличием коллагеновых волокон, встречается реже, чем вызванный ретикулиновыми нитями. Признаки остеомиелосклероза наблюдаются еще реже. Появление фиброза, переход от очагового к диффузному считается важным прогностическим фактором [9]. Выраженный фиброз обычно ассоциируется с более короткими сроками выживаемости. Но могут быть исключения. Описаны случаи ХМЛ, когда выраженный фиброз отмечался на ранних стадиях, но заболевание имело более длительное течение [10]. У ряда больных с Ph’-положительным ХМЛ могут быть выраженный фиброз за счет разрастания коллагеновых волокон и картина крови, подобная наблюдаемой при хроническом идиопатическом миелофиброзе. Поставить правильный диагноз в этих случаях помогает низкий уровень (или практически полное отсутствие) щелочной фосфатазы в нейтрофилах [1].
При ХМЛ крайне низкая активность лейкоцитарной щелочной фосфатазы (ЛЩФ) наблюдается во всех случаях — независимо от количества лейкоцитов в периферической крови и тяжести клинического течения заболевания [11]. Низкий уровень ЛЩФ обусловлен нарушением выработки фермента, а не дефектами его каталитической функции или нарушениями в образовании специфических гранул в цитоплазме лейкоцитов [12, 13]. После успешного лечения уровень ЛЩФ обычно возвращается к норме. Определение ЛЩФ, наряду с выявлением Ph’ хромосомы, используется в клинической практике для подтверждения диагноза в спорных и сомнительных случаях, когда ХМЛ приходится дифференцировать с рядом патологических процессов, сопровождающихся развитием лейкемоидных реакций миелоидного типа (инфекционные заболевания, метастазы опухолей, действие токсических веществ и др.).
К числу признаков, способствующих установлению диагноза ХМЛ (табл. 22), относится также повышенный уровень витамина В12 и витамин В12-связывающих белков в сыворотке крови больных, что, в свою очередь, обусловлено повышением содержания транскобаламинов I и II [14]. Как установлено, эти изменения, как и снижение активности ЛЩФ, увеличение количества базофилов и тромбоцитов, могут определяться за несколько лет до манифестацииХ МЛ [13].
В табл. 23 приведены клинико-лабораторные признаки, которые могут быть использованы в дифференциальной диагностике ХМЛ и лейкемоидных реакций миелоидного типа.
Иммунофенотипический анализ антигенов поверхностных мембран гранулоцитов в хронической фазе ХМЛ позволил обнаружить некоторые отличия по сравнению с клетками соответствующей степени зрелости в норме. Отмечено уменьшение плотности HLA-DR (Ia-подобного антигена) на трансформированных клетках-предшественниках гранулоцитарного ряда. На меньшем количестве клеток обнаруживался антиген CD13, увеличенным было число незрелых клеток с двойными маркерами CD15+ CD34+ [15–17], до 50% клеток реагировало с мкАТ к CD116 [17]. В целом, изучение маркеров поверхностных мембран клеток не играет существенной роли в диагностике ХМЛ в хронической фазе заболевания. При развитии же бластного криза иммунофенотипический анализ, наряду с данными цитохимических исследований, позволяет, как и при острых лейкозах, точнее идентифицировать природу бластных клеток, определяемых в костном мозге и периферической крови.
Цитогенетический анализ. Приблизительно у 5% больных с ХМЛ в лейкозных клетках при рутинном цитогенетическом исследовании не обнаруживается t (9; 22). Однако при использовании молекулярно-биологических методов и в этих случаях может быть обнаружен гибридный ген BCR/ABL. Образующийся при этом белок, обладающий повышенной активностью тирозинкиназы, может быть ответственным за усиленную пролиферацию клеток у больных с ХМЛ [2, 18–20].
Фаза акселерации и бластной трансформации
Хроническая фаза течения ХМЛ, средняя продолжительность которой составляет 3–4 года, сменяется более агрессивной и кратковременной фазой заболевания. У большинства больных развивается бластный криз (бластная трансформация), по клинико-гематологическим проявлениям близкий к острому лейкозу, развивающемуся de novo. Он характеризуется резистентностью к применяемой терапии и средней продолжительностью жизни от 3 до 6 мес [19–21]. У 10–30% больных с ХМЛ ухудшение состояния сочетается с нарастанием изменений в костном мозге и периферической крови. Но при этом количество бластов недостаточно для диагностики бластного криза. Эта стадия развития заболевания определяется как фаза акселерации, средняя продолжительность которой составляет от 12 до 18 мес [1, 3]. Сотрудниками Международного регистра по трансплантации костного мозга разработаны критерии, позволяющие четко определить переход в эти фазы заболевания [22].
Фаза бластной трансформации устанавливается при наличии не менее 30% бластов в костном мозге и периферической крови. Внезапное и быстрое увеличение содержания бластных клеток сопровождается нарастанием недостаточности костномозгового кроветворения, усилением выраженности анемии и тромбоцитопении. При бластном кризе у 70% больных патологические клетки имеют миелоидную природу, у 20–25% — лимфоидную. В 5% случаев бласты имеют дифференцировочные признаки ранних клеток-предшественников эритробластического или мегакариоцитарного ряда. Прогноз благоприятнее и чувствительность к терапии несколько выше при наличии бластов лимфоидного происхождения. Для более точного определения природы клеток при бластном кризе рекомендуется применение цитохимических и иммунологических методов [1, 2, 4, 5].
При бластной трансформации миелоидного типа клетки могут иметь цитоморфологические и цитохимические признаки миелобластов, монобластов, быть сходными с бластами при ОМЛ М4 и ОМЛ М3 [3].
При бластной трансформации лимфоидного типа клетки имеют цитологические признаки, присущие бластам при ОЛЛ L1 или L2 и очень редко ОЛЛ L3 [1, 3]. В большинстве случаев при бластном кризе лимфоидного типа клетки имеют В-клеточное происхождение и очень редко являются трансформированными ранними клетками-предшественниками Т ряда. При лимфоидном бластном кризе В-клеточного подтипа ранние клетки-предшественники имеют иммунофенотип ОЛЛ «общего типа» (ОЛЛ В II) или пре-В-клеточный (ОЛЛ В III). При бластном кризе ХМЛ лимфоидного типа чаще, чем при ОЛЛ, бластные клетки по данным иммунофенотипирования имеют бифенотипические/билинейные признаки [3]. Иногда в клинику поступают больные в стадии бластного криза, у которых по разным причинам ранее не был диагностирован ХМЛ. При этом бласты костного мозга и периферической крови могут иметь цитоморфологические и цитохимические признаки миелоидных или лимфоидных клеток-предшественников. Клинико-гематологическая картина в первом случае чаще всего напоминает ОМЛ М2, но при сохранении достаточно большого количества зрелых и незрелых гранулоцитов. При бластной трансформации лимфоидного типа не наблюдается признаков созревания TdT-положительных бластов в промиелоциты. Особые трудности возникают при дифференциальной диагностике лимфоидного бластного криза ХМЛ и Ph’-положительного ОЛЛ [1].
Почти у 30% больных с ХМЛ хроническая фаза сменяется фазой акселерации, предшествующей развитию бластного криза [1, 3]. Фаза акселерации характеризуется нарастанием миелофиброза, увеличением количества базофилов в периферической крови (>20%), снижением уровня гемоглобина (<7 г/дл). Но при этом количество бластов в костном мозге и периферической крови составляет менее 30% [21, 23]. Наблюдается также увеличение количества эозинофилов в крови и костном мозге (>10%), незрелых клеток моноцитарного ряда, эритробластов. В зрелых и незрелых миелоидных клетках обнаруживаются диспластические изменения: гиперсегментация ядер и гипогрануляция цитоплазмы нейтрофилов, различные признаки дисэритропоэза.
При исследовании трепанобиоптатов костного мозга также обнаруживаются черты дезорганизации и диспластические изменения. Увеличение количества гиподольчатых мегакариоцитов сопровождается усилением ретикулинового фиброза. Может наблюдаться коллагеновый фиброз, иногда сочетающийся с развитием остеосклероза. Увеличивается количество бластных клеток, располагающихся паратрабекулярно и периваскулярно, при одновременном уменьшении количества незрелых гранулоцитов.
Ряд больных с ХМЛ поступает в стационар в фазе акселерации, в таких случаях возникает необходимость проведения дифференциальной диагностики с различными формами МДС и острых лейкозов.
В фазе акселерации и при бластной трансформации у 80% больных с ХМЛ происходит дальнейшая эволюция кариотипа. Вторичные аномалии отмечаются чаще при миелоидном типе бластного криза, чем при лимфоидной трансформации. Основными дополнительными аномалиями, которые могут быть выявлены за несколько месяцев до развития бластного криза миелоидного типа, являются i(17q)+8, t (3; 21) (q26; q22), inv(3) (q21 q26), del(13 ) (q12 q14) [24]. Возникновение лимфоидного типа бластного криза наиболее часто сочетается с del(7) (q22) и –7.
Молекулярно-генетические аномалии, связанные с развитием бластного криза ХМЛ, касаются опухолеассоциированных супрессорных генов p53, RB1, p16 и реже онкогенов ras и myc [25, 26]. Мутации генов p16 и RB1 чаще наблюдаются в кроветворных клетках у больных с лимфоидным типом бластного криза, а гена p53 — при миелоидном типе бластной трансформации [25–27].
Атипичный миелопролиферативный синдром (Ph
–/BCR– миелолейкоз)Термины «атипичный миелопролиферативный синдром» (аМПС), «атипичный хронический лейкоз», «Ph’ отрицательный хронический миелолейкоз» употребляются для обозначения гетерогенной группы миелопролиферативных заболеваний. От ХМЛ аМПС отличается по ряду клинико-гематологических проявлений.
Основными признаками аМПС являются отсутствие Ph’-хромосомы и выявляемой реарранжировки генов BCR/ABL, гиперлейкоцитоз и нейтрофилез, наличие в периферической крови незрелых форм гранулоцитов. При аМПС, в отличие от ХМЛ, отмечаются диспластические изменения в клетках гранулоцитарного и эритробластического ряда. Количество базофилов — в пределах нормы или слегка повышено. Нечасто отмечается и эозинофилия. Количество лейкоцитов в периферической крови составляет от 20•109/л до 180•109/л. В лейкоцитарной формуле содержание промиелоцитов, миелоцитов и метамиелоцитов превышает 15%.
У больных с аМПС, а это в основном мужчины по возрасту на 15–20 лет старше, чем пациенты с ХМЛ, количество лейкоцитов в среднем ниже, но чаще наблюдается анемия и тромбоцитопения. Уровень гемоглобина колеблется в пределах от 3,4 до 14,2 г/дл. Анемия, наряду со спленомегалией, относится к числу основных жалоб при обращении за помощью к врачу-гематологу. Среднее количество тромбоцитов не превышает 80•109/л [1]. По сравнению с Ph’-положительным ХМЛ при аМПС в периферической крови увеличено относительное (но не более 10%) и абсолютное содержание моноцитов.
Диспластические изменения нейтрофилов проявляются неправильной формой ядер (псевдопельгеровские лейкоциты) и наличием гипогранулярной цитоплазмы у зрелых нейтрофилов, миелоцитов и метамиелоцитов. В большинстве случаев, как и при ХМЛ, снижен уровень активности ЛЩФ, хотя у отдельных больных может отмечаться повышенный показатель.
Костный мозг при аМПС, как правило, гиперклеточный за счет повышенного количества незрелых и зрелых клеток гранулоцитарного ряда. Но в отличие от ХМЛ лейко-эритроцитарное соотношение обычно менее 10:1. Нечасто увеличено количество незрелых базофилов и эозинофилов. Количество клеток моноцитарного ряда, напротив, увеличено. Количество мегакариоцитов у 30% больных уменьшено, в них, как и в клетках гранулоцитарного и эритробластического ряда, обнаруживаются диспластические признаки. Увеличено количество бластов, но оно не превышает 30%.
В терминальной стадии аМПС развивается бластный криз. Обнаруживаемые при этом в костном мозге и периферической крови бластные клетки чаще имеют миелоидную природу, реже относятся к клеткам-предшественникам лимфопоэза. Это служит еще одним доказательством того, что аМПС, как и ХМЛ, возникает в результате трансформации полипотентной стволовой кроветворной клетки.
Дифференциальную диагностику аМПС проводят с ХМЛ, различными формами МДС. Ряд сходных и перекрывающихся клинико-гематологических признаков имеют аМПС и такие миелопролиферативные процессы, как истинная полицитемия, эссенциальная тромбоцитемия и идиопатический миелофиброз в поздних стадиях заболевания.
Особенно сложной является дифференциация аМПС, ХМЛ и ХММЛ. ФАБ-группой предложены критерии, помогающие разграничить эти процессы [28]. Так, содержание базофилов в крови при аМПС и ХММЛ не превышает 2%, а при ХМЛ оно значительно выше. Количество моноцитов в крови больных с ХМЛ, как правило, ниже 3%, при аМПС — в пределах 3–10%, при ХММЛ — обычно выше 10%. Признаки дисплазии клеток гранулоцитарного ростка сильно выражены при аМПС, умеренно — при ХММЛ и практически не определяются при ХМЛ. Содержание незрелых гранулоцитов при аМПС колеблется в пределах 10–20%, при ХММЛ — ниже 10%, а при ХМЛ — выше 20%. Количество бластов в крови выше 2% в хронической фазе заболевания наблюдается только у больных с аМПС [1, 3]. Из числа цитогенетических аномалий при аМПС наиболее часто обнаруживается трисомия 8, моносомия 7 при отсутствии Ph’-хромосомы. Прогноз при аМПС значительно хуже, чем при ХМЛ и ХММЛ.
Хронический миеломоноцитарный лейкоз миелопролиферативного типа
Хронический миеломоноцитарный лейкоз — клональный процесс, обусловленный трансформацией полипотентной стволовой кроветворной клетки или клеток-предшественников, являющихся ее ближайшими потомками. Встречается преимущественно у лиц пожилого возраста, чаще у мужчин. Характеризуется совокупностью миелодиспластических и миелопролиферативных признаков. При преобладании первых, включая определяющуюся при исследовании периферической крови цитопению, заболевание классифицируется как одна из форм МДС — ХММЛ. Если заболевание по клинико-гематологическим проявлениям больше напоминает ХМЛ, оно обозначается как хронический миеломоноцитарный лейкоз миелопролиферативного типа (ХММЛ-МТ) [1]. Для данной формы заболевания присущи лейкоцитоз, нейтрофилез, повышенное содержание моноцитов (>3%) в периферической крови, наличие макроцитарной анемии и выраженной в различной степени спленомегалии. Абсолютное количество нейтрофилов в крови больных превышает 13•109/л, а моноцитов >1•109/л. В периферической крови обнаруживаются незрелые клетки нейтрофильного ряда. Обычно их содержание не превышает 10% от общего количества лейкоцитов. Бластные клетки встречаются редко. Изредка обнаруживают незрелые клеточные элементы моноцитарного ряда (промоноциты). Хотя это не является характерным признаком ХММЛ-МТ, но в сочетании с моноцитозом и отсутствием Ph’-хромосомы или гибридного гена BCR/ABL позволяет отличить данное заболевание от классического ХМЛ [29, 30]. Диспластические изменения в клетках гранулоцитарного ряда минимальные (псевдопельгеровские лейкоциты, нейтрофилы с гипогранулярной цитоплазмой и отрицательной реакцией при выявлении МПО) или отсутствуют. Уровень ЛЩФ у больных с ХММЛ-МТ снижен или находится в пределах нормы. Количество тромбоцитов обычно в пределах нормы или несколько уменьшено, в редких случаях отмечается тромбоцитоз.
При исследовании мазков из пунктатов отмечена гиперклеточность костного мозга с увеличением количества незрелых и зрелых клеток гранулоцитарного и в меньшей степени моноцитарного ряда. Последние составляют не менее 20% всех ядросодержащих кроветворных клеток. Клетки моноцитарного ряда представлены преимущественно моноцитами и небольшим количеством промоноцитов. Их наличие подтверждается при проведении цитохимических реакций на неспецифическую -НЭ и КНЭ [5]. При ХММЛ-МТ миелобласты и монобласты составляют не более 5% всех клеточных элементов костного мозга. Количество клеток эритробластического ряда различной степени зрелости и мегакариоцитов, как правило, в пределах нормы. Могут наблюдаться признаки дисгранулоцитопоэза и дисэритропоэза, появление гигантских многоядерных мегакариоцитов. При гистологическом изучении трепанобиоптатов костного мозга почти у 30% больных выявляется миелофиброз.
Определяемые приблизительно у трети больных цитогенетические аномалии (+8, –7 и del(20q), а также точечные мутации гена ras) не являются специфически ассоциированными с ХММЛ-МТ.
Дифференциальный диагноз ХММЛ-МТ проводят с ХМЛ, ХММЛ, лейкемоидными реакциями миелоидного типа и ОМЛ М4. Как известно, при ХММЛ-МТ в кроветворных клетках не обнаруживается Ph’ хромосома или гибридный ген BCR/ABL. Сложнее отличить ХММЛ-МТ от ХМЛ с диспластическими признаками, рассматривающегося в качестве одной из форм МДС. При этом учитывается количество лейкоцитов в периферической крови, наличие нейтрофилеза и моноцитоза, другие клинико-гематологические показатели, которые детально рассматривались выше. Проводя дифференциальную диагностику с лейкемоидными реакциями, особенно сопровождающимися моноцитозом, следует обращать внимание на их возможную связь с наличием опухолей или инфекционных заболеваний.
Течение заболевания характеризуется увеличением содержания незрелых клеток гранулоцитарного и моноцитарного ряда и прогрессирующей недостаточностью костномозгового кроветворения, возникновением осложнений, обусловленных развитием цитопении, или трансформацией в ОМЛ М4. Медиана выживаемости больных с ХММЛ-МТ ниже, чем у пациентов с ХМЛ.
Хронический нейтрофильный лейкоз
Хронический нейтрофильный лейкоз (ХНЛ) — редкое заболевание, встречающееся у лиц пожилого возраста (старше 60 лет), характеризуется наличием анемии, спленомегалии и иногда гепатомегалии. Диагноз ХНЛ устанавливают при проведении общего анализа крови, иногда случайно. Количество лейкоцитов в периферической крови колеблется в пределах 25–50•109/л и редко бывает выше 100•109/л. Преобладают сегментоядерные нейтрофильные лейкоциты (90–95%), но в отдельных случаях содержание палочкоядерных нейтрофилов составляет 20–50% [1, 3]. Крайне редко обнаруживаются незрелые гранулоциты (миелоциты, метамиелоциты) и ядросодержащие клетки эритробластического ряда, бласты отсутствуют. В некоторых нейтрофилах ядра имеют кольцевидную форму. В цитоплазме могут присутствовать токсическая зернистость и вакуоли. Миелодиспластические признаки (гипогрануляция цитоплазмы, псевдопельгеровские лейкоциты) не определяются. В лейкоцитах больных с ХНЛ, в отличие от ХМЛ, выявляется повышенный уровень щелочной фосфатазы. Морфологические признаки эритроцитов и тромбоцитов крови в пределах нормы. В сыворотке крови больных с ХНЛ, как и при ХМЛ, повышен уровень витамина В12 и витамин В12-связывающего белка.
При исследовании пунктатов и трепанатов костного мозга отмечается гиперклеточность, обусловленная выраженной гиперплазией нейтрофильных гранулоцитов. Представлены в основном зрелые и незрелые клеточные элементы этого ряда, количество миелобластов не увеличено. Признаки дисгранулоцитопоэза обычно отсутствуют. Может наблюдаться умеренное угнетение эритропоэза. Количество мегакариоцитов в пределах нормы или несколько повышено.
По цитохимическим и иммунофенотипическим признакам клетки крови и костного мозга не отличаются от соответствующих по степени зрелости гранулоцитов у здоровых людей. В кроветворных клетках при ХНЛ, в отличие от ХМЛ, не выявляются Ph’-хромосома и реарранжировка BCR/ABL. Описан ряд других цитогенетических аномалий, подтверждающих клональную природу ХНЛ, таких, как трисомия 8, трисомия 9, трисомия 21, 20q– и другие реарранжировки с вовлечением длинного плеча хромосомы 20 [31, 32].
О неопластической природе ХНЛ свидетельствуют также результаты исследований роста колоний в полутвердых средах. При этом гемопоэтические клетки-предшественники сохраняют ограниченную способность дифференцироваться исключительно в гранулоциты [33].
Важной является дифференциальная диагностика ХНЛ с нейтрофильными лейкемоидными реакциями, истинной полицитемией и хроническим идиопатическим миелофиброзом. Последние два заболевания могут также сопровождаться нейтрофилезом, а при ХНЛ в костном мозге некоторых больных могут отмечаться признаки фиброза и остеосклероза.
Прогноз при ХНЛ значительно хуже, чем у больных с ХМЛ. При прогрессировании ХНЛ в терминальный период в ряде случаев может происходить трансформация в ОМЛ [1, 3].
Гиперэозинофильный синдром/Хронический эозинофильный лейкоз
Диагноз гиперэозинофильного синдрома (ГЭС) или хронического эозинофильного лейкоза (ХЭЛ) очень трудно установить в ранней стадии заболевания. Подозрение на ГЭС/ХЭЛ возникает при наличии абсолютной эозинофилии (>1,5•109/л), удерживающейся на протяжении более 6 мес. При этом не удается установить причину эозинофилии (необходимо исключить заболевания, вызываемые паразитами, аллергию, коллагенозы, неходжкинские лимфомы, лимфогранулематоз, множественную миелому, метастазы). Развитие и созревание эозинофилов в костном мозге регулируется синергическим действием ГМ-КСФ, ИЛ-1, ИЛ-2, ИЛ-3, ИЛ-5 и другими эозинофилопоэтическими факторами, вырабатываемыми Т-лимфоцитами [2].
У больных с ГЭС/ХЭЛ могут обнаруживаться анемия, тромбоцитопения, гепатомегалия, спленомегалия, лимфаденопатия, нередко симптомы, обусловленные поражением сердца и легких. В таких случаях, если на основе цитогенетических и молекулярно-биологических исследований (включая и экспрессию гена N-ras) подтверждается клональность процесса, ставится диагноз ХЭЛ. При отсутствии этих данных диагноз ХЭЛ остается предположительным.
При исследовании периферической крови у больных общее количество лейкоцитов превышает 20•109/л. Отмечается персистирующая эозинофилия c преобладанием зрелых эозинофилов и небольшим количеством незрелых клеток. В некоторых случаях отмечаются гиперсегментация ядер, вакуолизация цитоплазмы клеток и уменьшенное содержание гранул. Аномальные гранулы или включения и палочки Ауэра не определяются. У части больных может быть умеренный нейтрофилез с небольшим количеством незрелых нейтрофилов.
Костный мозг представляется гиперклеточным за счет гиперплазии эозинофильного ростка. Представлены все стадии созревания эозинофилов, однако диспропорционального увеличения количества незрелых форм или бластов не наблюдается. В ряде случаев отмечается незначительное увеличение числа клеток нейтрофильного ряда. В эозинофилах при ХЭЛ, в отличие от нейтрофилов, не выявляется активность ХАЭ. Диспластически измененные бласты или незрелые клетки гранулоцитопоэза могут быть идентифицированы как эозинофилы на основании того, что в них определяется активность МПО, устойчивой к ингибированию цианидом. Эритропоэз и мегакариоцитопоэз не претерпевают существенных изменений.
В некоторых случаях при ХЭЛ обнаруживаются клональные хромосомные аномалии, такие как трисомия 8, i(17q), t (5; 12) (q31; q13), t (1; 5) (q23; q33) [34]. Дифференциальную диагностику проводят с Ph’ положительным ХМЛ с эозинофилией, с ХММЛ с эозинофилией, ассоциированным с t (5; 12) (q33; p13) и t (8; 13) (p11; q12), с ОЛЛ, изредка сопровождающимся эозинофилией, с ОМЛ М4Эо.
Хронический базофильный лейкоз
Базофильный лейкоз — редкое заболевание. Увеличение количества базофилов может наблюдаться в ряде случаев у больных с ХМЛ в хронической фазе и в фазе акселерации. Клетки с дифференцировочными признаками данного ряда могут наблюдаться при выделяемых в соответствии с ФАБ-классификацией подвариантах ОМЛ — М2Базо и М4Базо [3]. Базофильный лейкоз с содержанием бластов в костном мозге менее 30% имеет хроническое течение. В случаях обнаружения Ph’-хромосомы они должны рассматриваться как вариант ХМЛ [3]. При истинном хроническом базофильном лейкозе не выявляются Ph’-хромосома и реарранжировка BCR/ABL. Циркулирующие в периферической крови клетки содержат базофильные гранулы, дают отрицательную реакцию при выявлении активности МПО. В клинической картине высок удельный вес симптомов, связанных с освобождением больших количеств гистамина, наблюдается ДВС-синдром.
Хронический тучноклеточный лейкоз
Заболевание может возникнуть de novo или протекать в виде терминальной стадии системного мастоцитоза. Клиническое течение чаще хроническое или подострое, в периферической крови обнаруживаются немногочисленные тучные клетки (мастоциты). Эти атипичные клетки имеют дольчатые ядра, более высокое, чем в норме ядерно-цитоплазматическое соотношение, содержат в цитоплазме гранулы, которые метахроматически окрашиваются толуидиновым синим и дают интенсивную реакцию при цитохимическом выявлении активности ХАЭ. При исследовании мазков периферической крови могут определяться анемия, моноцитоз и эозинофилия. В эозинофилах отмечаются признаки дегрануляции и вакуолизации цитоплазмы. Костный мозг гиперклеточный за счет гиперплазии клеток гранулоцитарного ряда, эозинофилов и увеличенного количества лимфоцитов. В трепанобиоптатах костного мозга наблюдается очаговая или диффузная инфильтрация тучными клетками, гиперплазия клеток гранулоцитарного и мегакариоцитарного ряда, наличие миелофиброза и остеосклероза.
Исследования показывают, что злокачественно трансформированные тучные клетки имеют фенотип CD2+ CD4+ CD11b+ CD33+ CD8– CD19– TdT– HLA-DR–.
В клинической картине, помимо гепатомегалии, спленомегалии, лимфаденопатии, на первый план выступают симптомы, обусловленные повышенной выработкой гистамина.
Истинная полицитемия
Истинная полицитемия (ИП) (эритремия, синдром Вакеза — Ослера) — клональное миелопролиферативное заболевание, характеризующееся прежде всего избыточной продукцией клеток эритробластического ряда, а также гранулоцитов и мегакариоцитов. ИП — относительно редкое заболевание, ежегодно выявляется 5–10 новых больных на 1 млн населения. Соотношение мужчин и женщин составляет 1,2:1. Средний возраст, в котором впервые выявляется заболевание, составляет 60–70 лет. В то же время ИП иногда встречается в юношеском и даже в детском возрасте.
В своем развитии ИП проходит три последовательные стадии: фазу пролиферации, основным признаком которой является увеличение массы эритроцитов; фазу стабильного течения заболевания; фазу миелоидной метаплазии [35].
Основные критерии диагностики заболевания (табл. 24) были разработаны 25 лет тому назад группой по изучению ИП (PVSG) [36].
Диагноз ИП считается правомочным при наличии трех основных критериев категории А (А1+А2+А3) или двух первых критериев (А1+А2) и любых двух критериев категории В.
У некоторых больных для подтверждения диагноза могут использоваться и некоторые дополнительные признаки, такие, как низкий или нормальный уровень эритропоэтина в сыворотке крови (при повышенном уровне ИП исключается), рост эритроидных колоний в культуре в отсутствие эритропоэтина, клональность процесса, подтвержденная результатами цитогенетического и молекулярно-биологического исследования. Ценным является также гистологическое изучение трепанобиоптатов костного мозга, позволяющее подтвердить или исключить наличие ИП в сложных для диагностики случаях.
Симптомы, наблюдающиеся в начальной стадии заболевания, обусловлены увеличенной массой эритроцитов. Больные жалуются на слабость, головную боль, нередко в виде мучительной мигрени. У больных отмечается цианоз кожи и видимых слизистых оболочек (у 90%), боль в области сердца, в костях и нижних конечностях. У многих больных основной жалобой является кожный зуд. Нарушения со стороны ЦНС колеблются от легких функциональных (на основе стазов без тромбообразования) до тяжелых, обусловленных тромбозом крупных сосудов [37]. Тромбоз сосудов (почти у 30% больных) и кровоизлияния (у 25%) относятся к числу наиболее важных и грозных осложнений при ИП.
Одним из основных клинических симптомов у больных ИП, отнесенных к категории А, является увеличение размеров селезенки. Причиной спленомегалии, выявляющейся у 80% пациентов, является ее повышенное кровенаполнение и участие в лимфопролиферативном процессе. Почти у 70% больных одновременно обнаруживаются признаки гепатомегалии, обусловленные повышенным кровенаполнением органа, миелоидной метаплазией, разрастанием фиброзной ткани (в поздних стадиях заболевания).
Из данных лабораторных исследований основным для установления диагноза ИП является увеличение массы клеток красной крови. Присущее ИП увеличение количества эритроцитов (6–7 млн в 1 мм3) и уровня гемоглобина (18–22 г/дл) сопровождается повышением показателей гематокрита. В первой, «пролиферативной», стадии заболевания на нормобластическую эритроидную гиперплазию костного мозга указывает обнаружение в периферической крови нормохромных и нормоцитарных эритроцитов. При дефиците железа, обусловленном повышенной кровоточивостью или частыми лечебными кровопусканиями, в мазках крови могут выявляться гипохромные и микроцитарные эритроциты. В периферической крови более чем у 80% больных ИП отмечается нейтрофильный лейкоцитоз с небольшим сдвигом влево. Количество лейкоцитов обычно колеблется в пределах от 10 до 20•109/л. Умеренная базофилия наблюдается у 60–70% больных. Частой является эозинофилия.
Количество тромбоцитов повышено у 50–80% больных, у 10% из них оно выше 1000•109/л [37]. В периферической крови при ИП могут обнаруживаться крупные тромбоциты и фрагменты ядер мегакариоцитов.
В мазках костного мозга определяется гиперплазия за счет увеличения числа клеточных элементов не только нормобластического эритропоэза, но и клеток других ростков миелопоэза (гранулоцитарного, мегакариоцитарного).
Наличие гиперпластических процессов при ИП подтверждается и результатами гистологического изучения трепанобиоптатов костного мозга. У большинства больных отмечается гиперклеточность костного мозга. По данным исследователей, входящих в группу по изучению ИП, кроветворные клетки различного происхождения и разной степени зрелости занимают от 37 до 100% (в среднем 80%) всей площади срезов костного мозга [38]. Наиболее выражена гиперплазия клеток эритробластического ряда. Значительным является и увеличение количества мегакариоцитов, размер которых широко варьирует — от малых до очень крупных с дольчатыми ядрами [39]. Мегакариоциты в костном мозге распределяются в виде кластеров. В 25% случаев уже в начальной стадии ИП в костном мозге определяется увеличенное количество ретикулиновых волокон. В дальнейшем оно прогрессирует, что сочетается с увеличением клеточности костного мозга.
Длительность начальной и стабильной фаз ИП составляет от 5 до 20 лет. Примерно у 10–20% больных наблюдается переход в фазу миелоидной метаплазии. Для постполицитемической миелоидной метаплазии (ППММ) характерно уменьшение массы клеток красной крови, выявляемое при радиоизотопном исследовании, увеличение степени спленомегалии, усиление фиброза костного мозга с увеличением количества ретикулиновых и коллагеновых волокон, появление очагов экстрамедуллярного гемопоэза, развитие цитопении, пойкилоцитоз и анизоцитоз [1, 6]. Могут наблюдаться диспластические изменения в клетках эритробластического и гранулоцитарного ряда, не выявлявшиеся ранее. В ряде случаев они предшествуют развитию острого лейкоза, чаще всего миелоидного происхождения. Острый лейкоз возникает у 1–2% больных ИП, не лечившихся цитотоксическими препаратами, и у 10–15% пациентов после миелосупрессивной терапии [40]. В фазе ППММ риск развития острого лейкоза у больных значительно выше (23%), чем в начальной и стабильной фазе заболевания (7%).
Цитогенетические аномалии в момент установления диагноза выявляются почти у 40–50% больных ИП и ассоциируются с менее благоприятным прогнозом [1, 6]. Частота их увеличивается по мере прогрессирования заболевания, при эволюции в миелодиспластический процесс. Наиболее часто у больных ИП встречаются не являющиеся характерными только для этого заболевания следующие аномалии кариотипа: del(20q), +8, +9 [41]. Описания каких-либо особых изменений иммунофенотипа кроветворных клеток при ИП в доступной литературе отсутствуют.
Дифференциальную диагностику ИП проводят с другими заболеваниями, сопровождающимися вторичным (симптоматическим) эритроцитозом. Различают вторичные абсолютные эритроцитозы, при которых характерны раздражение эритробластического ростка костномозгового кроветворения и увеличение массы циркулирующих в крови эритроцитов, и относительные эритроцитозы, в основе которых лежит сгущение крови, обусловленное действием различных факторов. В онкогематологической клинике особую важность приобретает дифференциальная диагностика ИП в стабильной фазе заболевания и вторичных абсолютных эритроцитозов, встречающихся у больных с гипернефромой, опухолями почек, эндокринных органов.
ППММ и развивающийся острый лейкоз не создают диагностических проблем, так как возникают у больных ИП, длительно находившихся под наблюдением [1].
Эссенциальная тромбоцитемия
Эссенциальная тромбоцитемия (ЭТ) (синонимы: первичная тромбоцитемия, геморрагическая тромбоцитемия, идиопатическая тромбоцитемия) — миелопролиферативное заболевание, характеризующееся преимущественным поражением клеток мегакариоцитарного ряда, основным проявлением которого является выраженное увеличение количества тромбоцитов. Приводится ряд доказательств клональности процесса, обусловленного повреждением на уровне стволовой кроветворной клетки костного мозга или ее ближайших потомков [5, 42]. ЭТ относится к числу относительно редких заболеваний. Показатели ежегодной заболеваемости составляют 0,7 на 100 тыс. населения [43]. Заболевают люди в возрасте 50–70 лет, но описаны случаи развития ЭТ у лиц молодого возраста и даже у детей [44]. В клинической картине на первый план выступают осложнения, обусловленные тромбозом сосудов и кровоизлияниями. Активацией тромбоцитов и микротромбозами мелких сосудов обусловлены также часто наблюдающиеся у больных ЭТ неврологические проявления. У 50% больных определяется спленомегалия, а у 15–20% — гепатомегалия [45].
Количество тормбоцитов в периферической крови увеличено и нередко превышает 1000•109/л. Минимальным же для установления диагноза ЭТ в соответствии с критериями, предложенными группой экспертов по изучению ИП, является стойко удерживающийся в течение длительного времени уровень тромбоцитов, равный 600•109/л [46]. Уровень гемоглобина в соответствии с теми же критериями у больных ЭТ может колебаться в пределах от 10 до 18,8 г/дл (в среднем 13,8 г/дл). Среднее количество лейкоцитов в периферической крови составляет 11,5•109/л, но возможны значительные колебания — от 6 до 41•109/л. Незрелые клетки гранулоцитарного ряда в мазках периферической крови обнаруживаются крайне редко. Нет характерной для ХМЛ базофилии. Уровень щелочной фосфатазы, выявляемой при цитохимическом исследовании нейтрофилов, обычно находится в пределах нормы, в редких случаях может быть повышенным или сниженным.
Тромбоциты, выявляемые в мазках крови больных, не отличаются от наблюдающихся у здоровых людей, но могут быть полиморфными, варьируя по форме и величине. В ряде случаев могут обнаруживаться крупные, атипичные, гипогранулярные формы. Изредка в крови определяются ядросодержащие фрагменты мегакариоцитов, трудно отличимые от лимфоидных клеток. В этих случаях для идентификации природы клеточных элементов приходится прибегать к иммуноцитохимическому определению линейно-специфических антигенов — маркеров клеток мегакариоцитарного ряда, Т- и В-лимфоцитов.
У 70% больных при исследовании мазков из пунктатов и трепанобиоптатов обнаруживают гиперклеточный костный мозг. У отдельных больных количество миелокариоцитов может быть в пределах нормы или несколько увеличенным. Количество мегакариоцитов значительно повышено. Мегакариоциты в мазках равномерно распределены или образуют скопления в виде групп и кластеров. Размер мегакариоцитов с дольчатыми ядрами и обширной цитоплазмой нормальный или увеличенный. Обычно отмечается также умеренная гиперплазия клеток гранулоцитарного и эритробластического ряда. У 20–50% больных преимущественно пожилого возраста в костном мозге определяется увеличенное количество ретикулиновых волокон [46, 47].
В табл. 25 приведены критерии, позволяющие отличить ЭТ от других форм миелопролиферативных заболеваний (ИП, ХМЛ, идиопатического миелофиброза) и вторичных (реактивных) тромбоцитозов. Последние нередко отмечаются у больных с опухолями, воспалительными процессами, железодефицитной анемией, после операции спленэктомии. Следует отметить, что количество пластинок при вторичных тромбоцитозах редко превышает 1000•109/л.
Крайне сложной является дифференциальная диагностика ЭТ и ИП. Выраженный тромбоцитоз может быть также ранним проявлением ХМЛ. Лишь позднее увеличивается количество лейкоцитов и наблюдается выраженный сдвиг влево. В этих случаях помогает определение Ph’-хромосомы или обнаружение реарранжировки гена BCR/ABL.
Хронический идиопатический миелофиброз, или миелосклероз с миелоидной метаплазией, характеризуется более выраженной, чем при ЭТ спленомегалией. При гистологическом исследовании биоптатов костного мозга определяется фиброз с наличием сети коллагеновых волокон, а в крови — описанные ниже изменения.
Хронический идиопатический миелофиброз с миелоидной метаплазией
Хронический идиопатический миелофиброз с миелоидной метаплазией (ХИМММ) (синонимы; «миелосклероз с миелоидной метаплазией», «агногенная миелоидная метаплазия», «идиопатический миелофиброз», «алейкемический миелоз с остеосклерозом», «остеомиелосклероз и остеомиелофиброз») возникает в результате клональной пролиферации стволовых кроветворных клеток костного мозга [48]. Характерные признаки заболевания: панмиелоз, фиброз костного мозга и нередко остеосклероз, появление очагов экстрамедуллярного гемопоэза, спленомегалия, анемия, изменения лейкоцитарной формулы крови.
ХИМММ относится к числу редких форм миелопролиферативных заболеваний. Ежегодная заболеваемость составляет 0,5 на 100 тыс. населения [48]. Встречается преимущественно у лиц пожилого возраста (60–70 лет), но изредка и у детей [49]. Среди больных несколько преобладают лица мужского пола [50].
Основными жалобами больных при поступлении в клинику являются слабость, уменьшение массы тела, лихорадка, боль в костях, иногда геморрагические симптомы. Наиболее важными признаками ХИМММ считаются спленомегалия и гепатомегалия. В то же время у части больных первоначально отсутствуют какие-либо симптомы и заболевание выявляется случайно (на основе обнаружения увеличенной селезенки и результатов анализа периферической крови).
В момент установления диагноза может наблюдаться выраженная вариабельность гематологических параметров [51]. У большинства больных обнаруживают нормохромную анемию. У 50% больных уровень гемоглобина ниже 10 г/дл, а у 20% — 8 г/дл. Постоянными являются признаки анизоцитоза и пойкилоцитоза, наличие пойкилоцитов в форме слезинки (teardrop). У многих больных в мазках периферической крови обнаруживаются ядросодержащие клетки эритробластического ряда. Количество ретикулоцитов умеренно увеличено и может колебаться в зависимости от стадии заболевания. При наличии аутоиммунного гемолиза количество ретикулоцитов увеличивается до 5–20%.
Существенно варьирует содержание лейкоцитов в периферической крови. Изредка оно может быть низким, у 40% пациентов колеблется в пределах 10–25•109/л, у некоторых превышает 40•109/л. В крови у больных с ХИМММ встречаются гипер- или гипосегментированные лейкоциты, небольшой процент незрелых клеток гранулоцитарного ряда (миелоцитов и промиелоцитов). Уровень миелобластов, выявляемых у 40% больных, обычно не превышает 1–5%. Однако даже увеличение их количества до 10% еще не служит показателем перехода заболевания в более агрессивную фазу или трансформации в острый лейкоз [39]. Показатели лейкоцитарной щелочной фосфатазы, как правило, повышены, но у ряда больных может наблюдаться нормальный уровень или даже снижение ферментативной активности [49]. При исследовании мазков периферической крови больных ХИМММ отмечается умеренная базофилия. Количество моноцитов, как правило, не изменено. Нередко наблюдается относительная лимфоцитопения [18].
Количество тромбоцитов в периферической крови при ХИМММ может быть уменьшенным (менее 150•109/л), но может отмечаться и тромбоцитоз (более 500•109/л) [50]. В мазках периферической крови выявляются атипичные, крупные и гипогранулярные тромбоциты и аномальные мегакариоциты, в том числе микромегакариоциты, промегакариоциты и ядра мегакариоцитов. Прогрессирование заболевания сопровождается увеличением в крови количества этих клеток, нарастанием лейкоцитоза и повышением уровня незрелых клеток гранулоцитарного ряда.
Анализ миелограммы при изучении мазков костного мозга, полученных при стернальной пункции, не является показательным. Результаты разнятся в зависимости от того, попала ли игла в очаг миелоидной гиперплазии или в очаги фиброза и остеосклероза. Более информативным для установления диагноза ХИМММ, как и ряда других форм хронических миелопролиферативных заболеваний, является гистологическое изучение трепанобиоптатов костного мозга. При этом практически во всех случаях определяется гиперклеточность костного мозга, как правило, диффузная и реже очаговая. Синусы костного мозга расширены и заполнены пролиферирующими кроветворными клетками. Представлены клеточные элементы всех трех основных клеточных линий миелопоэза, хотя в отдельных срезах может быть преобладание клеток того или иного типа [1, 48]. У 90% больных отмечается выраженное количество мегакариоцитов, образующих кластеры из 3–10 клеток, характерно также наличие мегакариоцитов с конденсированными и дистрофически измененными ядрами в трепанобиоптатах с выраженным коллагеновым фиброзом.
Соотношение гемопоэтических клеток и фиброзной ткани варьирует не только в пробах, полученных из различных участков костного мозга, но и в срезах одного и того же биоптата [50]. Степень выраженности фиброза также значительно колеблется. В ранней стадии заболевания у большинства больных определяется увеличенное количество ретикулиновых волокон, располагающихся преимущественно вокруг сосудов [1, 51]. Коллагеновых волокон немного. При развитии интенсивного фиброза и возникновении очагов склероза клеточность костного мозга в отдельных участках снижается. Очажки сохранившихся клеточных элементов мегакариоцитарного, гранулоцитарного и эритробластического ряда разделены волокнами соединительной ткани. В некоторых участках костного мозга кроветворные клетки практически отсутствуют, фиброзная ткань целиком заполняет межтрабекулярные пространства.
Предпринимались попытки классификации и выделения отдельных типов ХИМММ с учетом результатов изучения трепанобиоптатов костного мозга. Так, Ward, Block [52] считают возможным выделение следующих трех основных гистологических форм: пангиперплазия, миелоидная атрофия и фиброз, миелофиброз и остеосклероз.
При пангиперплазии клетки трех основных линий миелопоэза занимают более 70% площади костного мозга. Иногда наблюдается увеличение числа ретикулиновых волокон при отсутствии коллагенового фиброза. Изредка встречаются скопления лимфоцитов. Подобная картина напоминает наблюдающуюся при ИП. При миелоидной атрофии и фиброзе небольшие очаги гемопоэза разделены ретикулиновыми и коллагеновыми волокнами. В их состав входят клетки всех основных ростков миелопоэза, но преобладают мегакариоциты. Увеличено количество плазматических клеток и клеток стромы. При третьей гистологической форме на первый план выступают признаки миелофиброза и остеосклероза. Количество гемопоэтических клеток уменьшено, и они представлены в основном мегакариоцитами.
Прогностически значимым может быть и разделение больных с ХИМММ на две группы с учетом данных гистопатологического изучения костного мозга при первоначальном установлении диагноза [51]. У больных первой группы (40%) костный мозг гиперклеточный с наличием атипичных мегакариоцитов, незрелых и зрелых гранулоцитов, клеток эритробластческого ряда, располагающихся в расширенных синусах. Ретикулиновые волокна отсутствуют или их количество несколько увеличено, располагаются они преимущественно вокруг сосудов. Коллагеновые волокна не выявляются. У больных второй группы (60%) в костном мозге отмечается умеренное или значительное уменьшение количества гемопоэтических клеток и очаги выраженного фиброза и склероза. Очажки, состоящие из незрелых клеток эритроидного и гранулоцитарного ряда, разделены участками соединительной ткани. Дистрофически измененные мегакариоциты, являющиеся преобладающими клеточными элементами, находятся в виде кластеров в расширенных синусах. У таких пациентов отмечается более низкий уровень гемоглобина и уменьшенное содержание тромбоцитов в крови, более выраженная степень спленомегалии, чем у больных первой группы.
Попутно заметим, что стромальные клетки, в первую очередь фибробласты, с которыми связывается развитие фиброза и остеосклероза, не относятся к популяции неопластических клеток. Представляется, что их пролиферация может быть связана с выработкой трансформированными мегакариоцитами повышенных количеств фактора роста фибробластов [1, 5].
Очаги экстрамедуллярного гемопоэза у больных с ХИМММ могут иметь различную локализацию, но наиболее часто возникают в селезенке, печени и лимфатических узлах [53]. В селезенке и печени клетки эритробластического, гранулоцитарного и мегакариоцитарного рядов располагаются в синусах. В лимфатических узлах, наряду с подобной же локализацией клеток трех основных ростков миелопоэза, может отмечаться выраженная перифолликулярная инфильтрация.
Цитогенетические аномалии наблюдаются почти у 60% больных с ХИМММ. Наиболее частой, хотя и неспецифической, является del 13 (q13q210). Другие возможные аномалии включают –7, +8 и +9. Кариотипические аномалии, выявляемые в момент установления диагноза, относятся к числу неблагоприятных прогностических признаков. В ряде случаев эволюция кариотипа ассоциируется с трансформацией в острый лейкоз.
Дифференциальную диагностику ХИМММ проводят со многими заболеваниями, сопровождающимися развитием фиброза и остеомиелосклероза. В первую очередь это различные формы злокачественных миелопролиферативных и лимфопролиферативных процессов. К числу первых относятся ХМЛ, ИП, ЭТ, острый миелофиброз. Для разграничения ХИМММ и ХМЛ применяют определение Ph’-хромосомы и перестройки BCR/ABL. Дифференциация ХИМММ и ИП крайне затруднена и во многих случаях должна основываться на анализе клинико-гематологических признаков.
При остром миелофиброзе отмечается агрессивное клиническое течение, отсутствие или небольшая степень спленомегалии. Помимо миелофиброза отмечаются признаки вовлечения в процесс основных ростков миелопоэза с увеличением количества незрелых и бластных клеток.
Развитием фиброза сопровождается поражение костного мозга также при неходжкинских злокачественных лимфомах. Но при этом, в отличие от ХИМММ, наблюдается мономорфная инфильтрация костного мозга клетками, соответствующими той или иной форме и цитологическому варианту исходных опухолей лимфоидной ткани.
Появление очагов специфического поражения при лимфогранулематозе также сопровождается разрастанием ретикулиновых и коллагеновых волокон. В дифференциальной диагностике решающим является обнаружение специфических для лимфогранулематоза гигантских многоядерных клеток Березовского—Штернберга.
Реактивный, или вторичный фиброз, наряду с остеосклерозом и остеолитическими изменениями, наблюдается и при метастатических поражениях костного мозга у больных раком молочной железы и других органов. В этих случаях помогает выявление раковых клеток в костном мозге на основе применения иммуноцитохимических методов и мкАТ к цитокератинам, онкофетальным антигенам, антигенам мембран эпителиальных клеток [4, 5, 54].
Развитие миелофиброза возможно также при коллагенозах, диссеминированном туберкулезе, действии ионизирующей радиации [4, 5]. Указанные заболевания и состояния также необходимо принимать во внимание при установлении диагноза ХИМММ.
Медиана выживаемости больных с ХИМММ составляет 3 года — 5 лет. Основными причинами смерти являются инфекции, кровоизлияния, тромбоэмболии. Трансформация в острый лейкоз, как правило, миелоидного происхождения отмечается у 5–20% больных [1, 48].
Хронический миелопролиферативный синдром, неклассифицируемый
Термин «хронический миелопролиферативный синдром, неклассифицируемый (ХМПС-Н)» был предложен группой по изучению ИП (PVSG) для обозначения заболевания у больных со спленомегалией, с варьирующими по степени тромбоцитозом и гиперплазией клеток мегакариоцитарного ряда, у которых не наблюдается увеличения массы эритроцитов, лейкоэритробластической реакции, значительного миелофиброза и отсутствует Ph’-хромосома [7]. Безусловно, некоторые случаи, диагностируемые как ХМПС-Н, могут быть ранними стадиями процесса, который в дальнейшем по мере получения достаточной информации удастся более точно классифицировать. Эту категорию не следует также использовать для обозначения случаев, при которых полученные при клинико-лабораторных исследованиях данные недостаточны для установления более точного диагноза.
Морфологические изменения в костном мозге и периферической крови весьма гетерогенны, но, вероятно, можно выделить следующие категории или подтипы.
Первый — случаи, когда при первоначальном исследовании костного мозга обнаруживается пангиперплазия с выраженной гиперплазией клеток мегакариоцитарного ряда, в связи с чем требуется проводить дифференциальную диагностику с ХИМММ или ИП и ЭТ. Но при этом отсутствуют обычные критерии, необходимые для установления диагноза указанных заболеваний. При исследовании периферической крови у таких больных определяют лейкоцитоз и/или выраженный тромбоцитоз. Количество эритроцитов не увеличено. Нет данных, указывающих на наличие очагов экстрамедуллярного гемопоэза.
Второй — случаи, когда начальные проявления напоминают ХИМММ (спленомегалия, лекоэритробластоз, пойкилоциты в виде слезинки, фиброз костного мозга), но при этом отмечаются выраженные диспластические изменения в клетках различных линий. Количество миелобластов существенно не повышено (<5%), в костном мозге происходит обычная дифференцировка клеток всех линий. Неясно, представляют ли такие случаи трансформацию хронической фазы ХИМММ в более агрессивное заболевание или являются «переходными» нарушениями с признаками миелопролиферативных и миелодиспластических процессов. Пока же их предлагается обозначать как ХМПС-Н.
Термин этот не может использоваться для обозначения патологических состояний с признаками, обычно наблюдающимися при хронических миелопролиферативных заболеваниях, такими, как пангиперплазия с выраженной гиперплазией мегакариоцитов, но при которых отмечаются выраженный сдвиг лейкоцитарной формулы влево с увеличением количества незрелых клеток и бластов (10–30%), миелофиброз и выраженная миелодисплазия. Последние, особенно при цитопении в периферической крови и отсутствии выраженной спленомегалии, скорее должны классифицироваться как миелодиспластические синдромы с миелофиброзом (включая «острую миелодисплазию с миелофиброзом», «острый миелосклероз» или «злокачественный миелосклероз»).
При ХМПС-Н, как и при других миелопролиферативных заболеваниях, обнаруживаются такие цитогенетические аномалии, как +8, +9, del(20q). Ph’-хромосома не определяется. Клинико-лабораторные данные у больных с ХМПС-Н вариабельны. Выделение прогностических признаков станет возможным после мониторинга течения процесса у достаточного числа больных.
Ювенильный хронический миелолейкоз (ЮХМЛ) не входит в классификацию хронических миелопролиферативных заболеваний, представленных в табл. 21. Это заболевание встречается исключительно у детей [55]. ЮХМЛ, наряду с синдромом моносомии 7, в качестве самостоятельной нозологической формы включен в классификацию миелодиспластических и миелопролиферативных заболеваний детского возраста. В ФАБ-классификации МДС [56] заболевание обозначается как ХММЛ. Этим же термином пользуются и участники Европейской рабочей группы по изучению МДС у детей [57, 58].
ЮХМЛ отличается от классического Ph’-положительного лейкоза, встречающегося в основном у взрослых, но иногда диагностируемого и у детей, по ряду морфологических, цитогенетических и клинических признаков [59, 60]. Это заболевание с агрессивным острым или подострым течением, напоминающим ОМЛ. Среди больных с ЮХМЛ 95% составляют дети (в основном мальчики) до 4 лет [55]. В момент установления диагноза возраст большинства детей не превышает 2 лет [57, 61]. Риск возникновения ЮХМЛ особенно высок у детей с семейным нейрофиброматозом [62].
Основными клиническими проявлениями заболевания являются анемия, гепатомегалия, спленомегалия, лимфаденопатия (у 20% больных), экземоподобные высыпания на коже головы, обусловленные лейкемическими инфильтратами.
У больных с ЮХМЛ умеренно снижен уровень гемоглобина. Уже в самом начале заболевания значительно уменьшается количество тромбоцитов. Общее количество лейкоцитов увеличено, но ниже, чем при ХМЛ у взрослых и колеблется в пределах 25–75•109/л [63]. При анализе лейкограммы определяют большой процент миелобластов, незрелых гранулоцитов, моноцитов, лимфоцитов и более низкий, чем при Ph’-положительном ХМЛ, уровень палочко- и сегментоядерных нейтрофильных лейкоцитов. Может определяться базофилия, не являющаяся, однако, постоянным признаком. В периферической крови могут обнаруживаться ядросодержащие клетки эритроидного ряда, количество которых увеличивается по мере прогрессирования заболевания. Встречаются отдельные плазматические клетки и иммунобласты. Костный мозг гиперклеточный, содержит увеличенное количество бластов (до 10–15%), незрелых и зрелых моноцитов. Содержание мегакариоцитов, как правило, уменьшено.
Уровень щелочной фосфатазы лейкоцитов, как и при Ph’-положительном ХМЛ, снижен. Важным лабораторным признаком является резкое увеличение (на 40–55%) уровня фетального гемоглобина у большинства больных по мере прогрессирования заболевания. При ХМЛ уровень фетального гемоглобина не превышает 10%. В качестве дополнительных признаков можно указать на наблюдающиеся при ЮХМЛ снижение гемоглобина А2 и активности карбоангидразы эритроцитов, увеличение активности глюкозо-6-фосфатдегидрогеназы и экспрессии антигена і при уменьшении экспрессии антигена І. У 50% больных повышен уровень сывороточных иммуноглобулинов [59].
У большинства больных при установлении диагноза ЮХМЛ не выявляют цитогенетических нарушений. Однако в ряде случаев описаны клональные аномалии хромосом (в основном трисомия 8), выявляемые в начальный период или в фазе прогрессирования заболевания. Частыми являются мутации онкогена ras [3].
Бластный криз развивается у небольшого числа детей. Но даже у больных, у которых не происходит бластная трансформация, прогноз неблагоприятный. Особенно тяжелое течение заболевания наблюдается у детей, заболевших в возрасте 6–12 мес. Медиана выживаемости больных с ЮХМЛ составляет 10–12 мес [3].
Дифференциальную диагностику ЮХМЛ проводят с такими заболеваниями, как редкий у детей Ph’ положительный ХМЛ, ОМЛ М4, синдром, связанный с моносомией 7, с другими миелодиспластическими/миелопролиферативными нарушениями.
1. Brunning RD, McKenna RW. Tumors of the bone marrow. Washington: Armed Forces Inst Pathol 1993; 406 p.
2. Schumacher HR, Coteligham JD. Chronic leukemia. Approach to diagnosis. New York: Igaku-Shoin Med Publ 1993; 356 p.
3. Bain BJ. Leukemia Diagnosis. 2nd ed. Oxford: Blackwell Science 1999. 200 p.
4. Hoffbrand AV, Pettit JE. Color atlas of hematology. 2nd ed. London etc: Mosby-Wolfe 1998. 360 p.
5. Глузман ДФ, Абраменко ИВ, Скляренко ЛМ, Надгорная ВА. Лабораторная диагностика онкогематологических заболеваний. Киев: Морион 1998. 336 с.
6. Neoplastic Hematopathology. Ed by DM Knowles, Baltimore et al: Williams a Wilkins 1992; 1624 p.
7. Imbert M, Pierre R, Vardiman J. Chronic Myeloproliferative Disorders. Writting Committee. Chicago 1998; 1–12.
8. Morrison VA. Chronic leukemias. Ca-A Cancer J Clin 1994; 44: 353.
9. Dekmezian R, Kantarjian HM, Keating MY et al. The relevance of reticulin stain-measured fibrosis at diagnosis in chronic myelogenous leukemia. Cancer 1987; 59: 1739.
10. Clough V, Geary CG, Hashmi K et al. Myelofibrosis in chronic granulocytic leukaemia. Br J Haematol 1979; 42: 515.
11. Глузман ДФ. Диагностическая цитохимия гемобластозов. Киев: Наук думка 1978; 216 с.
12. Wachstein M. Histochemistry of enzymes in tumors. Handbuch der Histochemie, Bd 2. 1962: 73.
13. Kamada N, Uchino H. Chronologic sequence in appearance of clinical and laboratory findings characteristic of chronic myelocytic leukemia. Blood 1978; 51: 843.
14. Zittoun J, Zittoun R, Marquet J, Sultan C. The three transcobalamins in myeloproliferative disorders and acute leukaemia. Br J Haematol 1975; 31: 287.
15. Todd MB, Waldron JA, Jennings TA et al. Loss of myeloid differentiation antigens precledes blastic transformation in chronic myelogenous leukemia. Blood 1987; 70: 122.
16. Schmetzer HM, Gerhartz HH. Immunological classification of chronic phase imminent blastic transformation and acute lymphoblastic leukemia. Exp Hematol 1997; 6: 502.
17. Логинский ВЕ, Выговская ЯИ, Потемкина ГИ и др. Иммунофенотипирование мононуклеарных клеток при хронических миелопролиферативных заболеваниях. Эксперим онкология 1991; 13: 37.
18. Wardiman JW. Chronic myelogenous leukemia and myeloproliferative disorders. In: Neoplastic Hematopathology 1992: 1405.
19. Lichtman MA. Chronic myelogenous leukemia and related disorders. In: Williams Hematology. 5th ed. 1995: 298.
20. Deininger MW, Goldman JM. Chronic myeloid leukemia. Curr Opin Hematol 1998; 5: 302.
21. Katarjian HM, Deisseroth A, Kurzrock R et al. Chronic myelogenous leukemia: a coincise update. Blood 1993; 82: 691.
22. Savage DG, Szydlo RM, Chase A et al. Bone marrow transplantation for chronic myeloid leukemia: the effects of differing criteria for defining chronic phase on probabilities of survival and relapse. Br J Haematol 1997; 99: 30.
23. Muehleck SD, McKenna RW, Arthur DC et al. Transformation of chronic myelogenous leukemia: clinical, morphologic and cytogenetic features. Am J Clin Pathol 1984; 82: 1.
24. Champlin RE, Golde DW. Chronic myelogenous leukemia: recent advances. Blood 1985; 65: 1039.
25. Ahuja HG, Jat PS, Foti A et al. Abnormalities of the retinoblastoma gene in the pathogenesis of acute leukemia. Blood 1991; 78: 3259.
26. Ishikura H , Yufu Y, Yamashita S et al. Biphenotypic blast crisis of chronic myelogenous leukemia: abnormalities of p53 and retinoblastoma genes. Leuk Lymphoma 1997; 25: 573.
27. Sill H, Goldman JM, Gross NCP. Homozygous deletion of the p16 tumor-suppressor gene are associated with lymphoid transformation of chronic myeloid leukemia. Blood 1995; 85: 2013.
28. Bennet JM, Catovsky D, Daniel MT et al. The chronic myeloid leukemias: guidelines for distinguishing chronic granulocytic, atypical myeloid, and chronic myelomonocytic leukemia. Proposals by the French-American-British Cooperative group. Br J Haematol 1994; 87: 746.
29. Pugh WC, Pearson M, Vardiman JW, Rowley JD. Philadelphia chromosome-negative chronic myelogenous leukaemia: a morphologic reassessment. Br J Haematol 1985; 60: 457.
30. Kantarjian HM, Kurzrock R, Talpaz M. Philadelphia chromosome-negative chronic myelogenous leukemia and chronic myelomonocytic leukemia. Hematol Oncol Clin North Am 1990; 4: 389.
31. Hasle H, Olesen G, Kerndrup G et al. Chronic neutrophilic leukemia in adolescence and young adulthood. Br J Haematol 1996; 94: 628.
32. Matano S, Nakamura S, Kobayashi K et al. Deletion of the long arm of chromosome 20 in a patient with chronic neutrophilic leukemia: cytogenetic findings in chronic neutrophilic leukemia. Am J Hematol 1997; 54: 72.
33. Yanagisava K, Ohminami H, Sato M et al. Neoplastic involvement of granulocytic-monocytic, or erythrocytic lineage in a patient with chronic neutrophilic leukemia. Am J Hematol 1998; 57: 221.
34. Bain BJ. Eosinophilic leukaemias and the idiopathic hypereosinophilic syndrome. Br J Haematol 1996; 95: 2.
35. Beutler E. Polycythemia vera. In: Williams Hematology. 5th ed. 1995; 324.
36. Berlin NJ. Diagnosis and classification of the polycythemias. Semin Hematol 1975; 12: 339.
37. Berk PD, Goldberg JD, Donovan PB et al. Therapeutic recommendation in polycythemia vera based on Polycythemia Vera Study Group protocols. Semin Hematol 1986; 23: 132.
38. Ellis JT, Peterson P, Geller SA, Rappaport H. Studies of the bone marrow in polycythemia vera and the evolution of myelofibrosis and secondary hematologic malignancies. Semin Hematol 1986; 23: 144.
39. Vykoupil KF, Thiele J, Stangel W et al. Histopathology, ultrastructure and cytogenetics of the bone marrow in comparison with secondary polycythemia. Virchovs Arch A 1980; 389: 307.
40. Landaw SA. Acute leukemia in polycythemia vera. Semin Hematol 1986; 23: 156.
41. Diez-Martin JL, Graham DL, Petitt RM et al. Chromosome studies in 104 patients with polycythemia vera. Mayo Clin Proc 1991; 66: 287.
42. Fialkow PY, Faquet GB, Jacobson RJ et al. Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood 1981; 58: 916.
43. McIntyre KJ, Hoagland HC, Silverstein MN, Petitt RM. Essential thrombocythemia in young adults. Mayo Clin Proc 1991; 66: 149.
44. Mitus AJ, Schsfer AJ. Thrombocytosis and thrombocythemia. Hematol Oncol Clin North Am 1990; 4: 157.
45. Schafer AJ. Essential (primary) trombocythemia. In: Williams Hematology 5th ed. 1995; 340.
46. Murphy S, Iland H, Rosental D, Laszlo J. Essential thrombocythemia: an interim report from the Polycythemia Vera Study Group. Semin Hematol 1986; 23: 177.
47. Wintrobes Clinical Haematology, 10th ed. Baltimore: Williams a Wilkins 1999; 2764 p.
48. Lichtman MA. Idiopathic myelofibrosis (agnogenic myeloid metaplasia). In: Williams Hematology 5th ed. 1995; 311.
49. Weinstein IM. Idiopathic myelofibrosis: Historical review, diagnosis and management. Blood Rev 1991; 5: 98.
50. Visani G, Finelli C, Castelli U et al. Myelofibrosis with myeloid metaplasia: clinical and haematological parameters predicting survival in a series of 133 patients. Br J Haematol 1990; 75: 4.
51. Thielle J, Zankovich R, Steinberg T et al. Agnogenic myeloid metaplasia (AMM) — correlation of bone marrow lesions with laboratory data: a longitudinal clinicopathological study of 114 patients. Hematol Oncol 1989; 7: 327.
52. Ward HP, Block MH. The natural history of agnogenic myeloid metaplasia (AMM) and a critical evaluation of its relationship with myeloproliferative syndrome. Medicine 1971; 50: 357.
53. Beckman EN, Oehrle JS. Fibrous hematopoietic tumors arising in agnogenic myeloid metaplasia. Hum Pathol 1982; 13: 804.
54. Cripe LD, Hromas RW. Malignant disorders of megakaryocytes. Semin Hematol 1998; 35: 200.
55. Passmore SJ, Hann JM, Stiller CA et al. Pediatric myelodysplasia: a study of 68 children with a new prognostic scoring system. Blood 1995; 85: 1742.
56. Bennet JM, Catovsky D, Daniel MT et al. Proposed revised criteria for the classification of acute myeloid leukemia. Ann Intern Med 1985; 103: 626.
57. Neimeyer CM, Arico M, Basso G et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. Blood 1997; 89: 3534.
58. Neimeyer CM, Fenu S, Hasle H et al. Differentiating juvenile myelomonocytic leukemia from infections disease. Blood 1998; 91: 365.
59. Smith KL, Johnson W. Classification of chronic myelocytic leukemia in children. Cancer 1974; 34: 670.
60. Freedman MH, Estrov Z, Chan HS. Juvenile chronic myelogenous leukemia. Am J Pediatr Hematol Oncol 1988; 10: 261.
61. Arico M, Biondi A, Pui CH. Juvenile myelomonocytic leukemia. Blood 1997; 90: 479.
62. Clark RD, Hutter JJ. Familial neurofibromatosis and juvenile chronic myelogenous leukemia. Hum Genet 1982; 60: 230.
63. Hardisty RM, Speed DE, Till M. Granulocytic leukemia in childhood. Br J Haematol 1964; 10: 551.
Лейкоз:Причины,Симптомы,Лечение | doc.ua
В случае лейкемии происходит выработка измененных лейкоцитов, которые не отмирают при изнашивании или после получения повреждений. Такие клетки вытесняют нормальные клетки крови.
Классификация
Лейкемию можно классифицировать несколькими методами. В основном лейкоз крови классифицируют согласно характеру протекания болезни и типу лейкоцитов, подвергшихся поражению.
Первый критерий позволяет выделить такие виды лейкемии как острый и хронический лейкоз. Отличительной особенностью этого заболевания является невозможность перехода от острой формы к хронической.
При развитии хронического лейкоза наблюдается частичное выполнение функций пораженными лейкоцитами бессимптомное протекание лейкоза. Чаще всего обнаружить хроническую лейкемию можно во время осмотра у врача до момента проявления симптомов.
С течением времени происходит проявление симптоматической картины заболевания, вследствие увеличения количества лейкозных клеток. В основном болезнь проявляется посредством инфекций и увеличенных лимфоузлов. С возрастанием поражения проявляются более серьезные признаки лейкемии.
Острая форма лейкоза отличается высокой скоростью распространения незрелых клеток, которые не могут выполнять свои функции. Развитие заболевания происходит стремительно.
Другой метод классификации основан на типе белых клеток крови, подверженных процессу озлокачествления. Выделяют такие виды заболевания как острый лимфобластный лейкоз, острый миелобластный лейкоз либо острый миелоидный лейкоз.
Существует четыре группы лейкоза, позволяющие охватить практически все его виды.
Хронический лимфоцитарный лейкоз
Медленно протекающая форма, при которой поражаются лимфоидные клетки организма. В большинстве случаев пациентами становятся люди возрастом от 55 лет. Лейкоз у детей не наблюдается в данной категории. Результат прогноза пятилетней выживаемости составляет 75%.
Хронический миелобластный лейкоз
Наблюдается данный лейкоз у взрослых, в большинстве случаев – у людей пожилого возраста. Поражению подвергаются миелоидные клетки. Ежегодное количество случаев заболевания превышает 5000 человек. Пятилетняя выживаемость – 90%.
Острый лимфобластный лейкоз
Быстро прогрессирующее поражение лимфоидных клеток. Каждый год фиксируется более 5 тысяч случаев болезни. Данный вид лейкоза является наиболее распространенным видом лейкоза среди детей и взрослых. Также может возникнуть у людей пожилого возраста. Прогноз выживаемости составляет не более 50% для взрослых и около 90% у детей.
Острый миелоидный лейкоз
Агрессивное распространение раковых клеток миелоидного типа. Больными могут быть как взрослые, так и дети. Ежегодное число заболеваний составляет примерно 13000 случаев. Прогноз выживаемости не превышает 40%.
Кроме того, обособленными видами лейкоза могут быть виды заболевания, которые встречаются реже остальных, к примеру, волосатоклеточный лейкоз либо лейкоз врх.
Причины
Ученые определили определенные факторы, способствующие развитию данного заболевания.
К факторам риска лейкоза относятся
- радиоактивное облучение, провоцирующих развитие миелобластных лейкозов острой и хронической формы, а также острого лимфобластного лейкоза;
- радиоактивные выбросы;
- лучевая терапия в случае частого ее применения;
- рентгенография, увеличивающая риск развития лейкемии при частом применении во время различных обследований;
- курение;
- взаимодействие с реагентами химического типа, в частности, бензолом;
- применение химиотерапии;
- наследственные заболевания, к примеру, синдром Дауна;
- заболевания крови, к примеру, миелодиспластический синдром.
Однако стоит учитывать, что наличие одного или нескольких факторов из группы риска, не во всех случаях приводит к развитию у человека лейкоза. Однако рекомендуется избегать контакта с этими факторами, чтобы предупредить развитие лейкемии в будущем.
Симптомы
Симптоматическая картина данного заболевания зависит от типа лейкоза, места скопления и количества пораженных клеток. В случае повреждения тканей центральной нервной системы могут возникать спутанное сознание, потеря мышечного контроля, рвота, постоянная головная боль.
Влияние лейкемии может распространяться на функционирование внутренних органов, к примеру, почек, легких, сердца, что становится причиной появления симптомов нарушенной работы этих органов.
Симптомы, общие для острой и хронической форм лейкоза, включают в себя
- увеличенные лимфатические узлы подмышек, паха, шеи, не причиняющие неудобств пациенту;
- беспричинные повышения температуры тела и повышенное выделение пота в ночное время;
- частое возникновение заболеваний инфекционного характера;
- высокий уровень слабости и утомляемости;
- возникающие кровоподтеки либо кровотечения;
- вздутие живота либо появление болезненных ощущений в области живота вследствие увеличения объемов внутренних органов, расположенных в этой области;
- потеря аппетита и веса;
- болезненные ощущения в суставах и костях.
Данные симптомы и признаки не могут однозначно указать на наличие лейкоза у человека. По этой причине в случае проявления любого из перечисленных признаков необходимо пройти диагностику незамедлительно.
Диагностика
Постановка точного диагноза возможна исключительно после осмотра врача и прохождения ряда процедур диагностического характера.
При осмотре врача применяется пальпация лимфатических узлов, печени и селезенки.
Проведение полного анализа крови устанавливает количество клеток в крови, что позволяет определить наличие лейкоза.
Наиболее достоверным методом диагностики лейкоза является взятие у больного биопсии. Это позволит установить наличие в организме раковых клеток. Проведение процедуры биопсии осуществляется исключительно при местном обезболивании. Используется костный мозг тазовых костей либо других больших костей организма пациента. Ткани берутся с применением тонких либо толстых игл.
В некоторых случаях допустимо применение дополнительных диагностических процедур
- цитогенетики – исследования хромосом кровяных клеток, клеток лимфоузлов либо костного мозга пациента;
- проведение исследования жидкости спинного мозга – данный метод диагностики проводится под воздействие местной анестезии и длится не более 30 минут;
- рентгенография области грудной клетки – позволяет зафиксировать лимфоузлы, подвергшиеся увеличению либо другие симптомы развития заболевания в области груди пациента.
Лечение
Результаты проведенных анализов крови и биопсии позволяет определить вид развивающегося лейкоза. Распространенность опухолевого процесса дает возможность вычислить стадию заболевания. Согласно этим сведениям происходит назначение адекватного лечения.
Основным методом лечения лейкоза является проведение химиотерапии. В данном методе применяются лекарственные препараты, склонные к разрушению раковых клеток, в некоторых случаях — замедляющие их размножение, чем приостанавливают распространение опухоли по организму человека. Применяться могут как один, так и несколько препаратов.
Выделяют несколько способов проведения химиотерапии, а именно: пероральный способ, внутривенное введение препаратов, интратектальная химиотерапия, введение препаратов в спинномозговую жидкость, введение в кожу головы.
Применение интратектальной химиотерапии дает некоторые преимущества по сравнению с другими методами. Использование этого метода позволяет препаратам проникнуть в узкие сосуды головного и спинного мозга.
Курсы лечения с применением химиотерапии проводятся с перерывом от 2 до 4 недель. Проведение возможно в отделении либо же в амбулаторных условиях, исходя из сложности процесса лечения, состояния пациента, побочных факторов и т.д. Длительность процесса лечения способна меняться.
Побочные эффекты химиотерапии
Вследствие замедленного размножения и роста нормальных клеток организма может возникать кровотечение, рвота, выпадение волос. В некоторых случаях применение химиотерапии приводит к развитию бесплодия. У мужчин прекращается выработка сперматозоидов, а женщинам грозит повреждение яичников.
В связи с этим мужчинам рекомендуется сдать семя до начала проведения лечения, которое будет заморожено. Женщинам же необходимо изъять яйцеклетку до начала применения химиотерапии. Данные меры предпринимаются в том случае, если пациент имеет желание иметь детей в будущем.
Также при лечении лейкоза могут использоваться целевая терапия, лучевая терапия, пересадка костного мозга, иммунотерапия и наблюдение. Также допустимо применение оперативного вмешательства по удалению селезенки в случае чрезмерного увеличения органа.
Публикации в СМИ
Лейкоз (лейкемия) — системное заболевание крови, характеризующееся замещением нормального костномозгового кроветворения пролиферацией менее дифференцированных и менее функционально активных клеток — ранних предшественников клеток лейкоцитарного ряда.
Классификация. По течению заболевания различают хронический и острый лейкозы • Острый. Без лечения приводят к смертельному исходу в течение недель или месяцев. Если проводить лечение, прогноз для детей часто благоприятен. Морфологический субстрат — бластные клетки • Хронический. Больные живут без лечения в течение нескольких месяцев и лет. Хронические лейкозы трансформируются в неподдающиеся лечению острые формы (бластный криз). Морфологический субстрат — лейкемические клетки, более дифференцированы и функционально активнее бластных клеток. Выделяют •• Хронический миелолейкоз •• Хронический лимфолейкоз и волосатоклеточный лейкоз •• Полицитемия истинная •• Остеомиелофиброз •• Парапротеинемические гемобластозы, характеризующиеся секрецией патологического белка парапротеина •• Миелома множественная •• Макроглобулинемия Вальденстрёма •• Болезнь тяжёлых цепей.
Факторы риска • Ионизирующее излучение • Воздействие химических (в т.ч. лекарственных) веществ •• Бензол •• Продукты перегонки нефти •• Цитостатики и некоторые другие ЛС • Иммунодефициты • Вирусы (например, вирус Эпстайна–Барр) • Наследственные хромосомные дефекты (различные транслокации, инверсии, делеции и т.д.).
Патогенез обусловлен нарушением синтеза ДНК кроветворной клетки, изменением генетического кода, бесконтрольными ростом и дифференцировкой определённого клона кроветворных клеток. Особенность — постепенное прогрессирование опухолевого процесса (опухолевая прогрессия). Проявления • Угнетение нормального кроветворения • Смена дифференцированных клеток недифференцированными • Лейкемизация (инфильтрация бластными клетками) других некроветворных органов • Резистентность лейкозных клеток к химиотерапии • Гетерогенность лейкозных клеток в разных очагах пролиферации.
МКБ-10 • C90.1 Плазмоклеточный лейкоз • C91 Лимфоидный лейкоз [лимфолейкоз] • C92 Миелоидный лейкоз [миелолейкоз] • C93 Моноцитарный лейкоз • C94 Другой лейкоз уточнённого клеточного типа • C95 Лейкоз неуточнённого клеточного типа • C96 Другие и неуточнённые злокачественные новообразования лимфоидной, кроветворной и родственных им тканей
Примечание. Хронический миелолейкоз, полицитемию истинную, остеомиелофиброз, а также тромбоцитоз идиопатический относят к миелопролиферативным заболеваниям, характеризующимся гиперплазией миелоидной ткани.
Приложение. Лейкоз волосатоклеточный — редкое заболевание, относящееся к группе хронических лейкозов и характеризующееся пролиферацией «волосатых» клеток (предположительно В-лимфоцитов) в органах ретикулоэндотелиальной системы и в крови. Характерны постепенное начало заболевания, спленомегалия, панцитопения, незначительная лимфаденопатия. При исследовании мазков: поверхность злокачественных клеток имеет многочисленные микроворсинки. Лечение • Спленэктомия • Интерферон. Синоним: лейкемический ретикулёз. МКБ-10. C95 Лейкоз неуточнённого клеточного типа
РАЗНИЦА МЕЖДУ ОСТРЫМ И ХРОНИЧЕСКИМ ЛЕЙКОЗОМ | СРАВНИТЕ РАЗНИЦУ МЕЖДУ ПОХОЖИМИ ТЕРМИНАМИ — ЖИЗНЬ
Острый и хронический лейкоз Лейкемия — это разновидность рака клеток крови. Есть четыре типа лейкемии; два типа острого лейкоза и два типа хронического лейкоза. Двумя острыми лейкозами являются остры
Острый и хронический лейкоз
Лейкемия — это разновидность рака клеток крови. Есть четыре типа лейкемии; два типа острого лейкоза и два типа хронического лейкоза. Двумя острыми лейкозами являются острый лимфобластный лейкоз (ОЛЛ) и острый миелоидный лейкоз (ОМЛ). Двумя хроническими лейкозами являются хронический лимфолейкоз (ХЛЛ) и хронический миелоидный лейкоз (ХМЛ). Большинство лейкозов вызываются специфическимигенетические мутации, делеции или транслокации. Все они имеют похожие симптомы и признаки; однако они требуют разных методов лечения. В этой статье будут рассмотрены все четыре типа лейкемии и различия между ними, выделены их клинические особенности, причины, исследования и диагноз, прогноз, а также различные методы лечения, необходимые для каждого из них.
Острый лейкоз
Острый лимфобластный лейкоз (ОЛЛ) проявляется как неопластическое разрастание лимфобластов (незрелыхлимфоциты). Классификация ВОЗ делит ОЛЛ на В-лимфоцитарный лейкоз и Т-лимфоцитарный лейкоз. Иммунологически ОЛЛ классифицируется как Т-клеточный ОЛЛ, В-клеточный ОЛЛ, Нуль-клеточный ОЛЛ и обычный ОЛЛ. Их симптомы и признаки связаны с поражением костного мозга. Низкийгемоглобин, инфекции, кровотечение, боль в костях, воспаление суставов, увеличение селезенки,лимфатический узел увеличениевилочковая железа расширение, ичерепно-мозговой нерв параличи — общие черты ALL. Опоясывающий лишай, ЦМВ, корь и кандидоз — частые инфекции, наблюдаемые у ВСЕХ пациентов. Предотвращение инфекций с помощьюантибактериальная терапия и вакцинация, химиотерапия для индукции ремиссии, консолидации и поддержания ремиссии являются важными шагами в лечении ОЛЛ. Трансплантация костного мозга также играет важную роль в лечении ОЛЛ.
Острый миелоидный лейкоз (ОМЛ) представляет собой неопластическую пролиферацию, происходящую из миелоидных элементов костного мозга. Это очень быстро прогрессирующая злокачественная опухоль. Существует пять типов ПОД. Это ОМЛ с генетическими аномалиями, ОМЛ с дисплазией нескольких линий, миелодиспластический синдром ОМЛ, ОМЛ неоднозначного происхождения и неклассифицированный ОМЛ. Анемия, инфекция, кровотечение, диссеминированное внутрисосудистое свертывание, боль в костях, сдавление пуповины, большая печень, большая селезенка, увеличение лимфатических узлов, недомогание, летаргия и боль в суставах являются общими признаками ОМЛ. Поддерживающая терапия, такая как переливание крови, антибиотики, химиотерапия и трансплантация костного мозга, являются обычными методами лечения.
Хронический лейкоз
Хронический миелоидный лейкоз (ХМЛ) характеризуется неконтролируемым разрастанием миелоидных клеток. На его долю приходится 15% лейкозов. Это миелопролиферативное заболевание, имеющее общие черты с этими заболеваниями. Общие признаки — потеря веса, подагра, лихорадка, потливость, кровотечение и боль в животе, анемия, большие размеры печени и селезенки. Филадельфийская хромосома — гибридная хромосома, образованная после транслокации хромосома 9–22. Обычно используются мезилат иматиниба, гидроксимочевина и аллогенная трансплантация.
Хронический лимфолейкоз (ХЛЛ) представляет собой моноклональное разрастание малых лимфоцитов. Пациенту обычно больше 40 лет. Мужчины поражаются вдвое чаще, чем женщины. ХЛЛ составляет 25% лейкозов. Это приводит к аутоиммунному гемолизу, инфекции и отказу костного мозга. Для лечения ХЛЛ необходимы лучевая терапия, химиотерапия и поддерживающая терапия.
В чем разница между острым и хроническим лейкозом?
• Острые лейкозы — это злокачественные опухоли незрелых клеток, а хронические лейкозы — злокачественные опухоли зрелых клеток.
• Острые лейкозы чаще встречаются у молодых людей, тогда как хронические лейкемии распространены у пожилых людей.
• Каждый тип лейкемии требует разных методов лечения.
Читать далее:
1. Разница между раком костей и лейкемией
2. Разница между лейкемией и лимфомой
3. Разница между лейкемией и миеломой
Различия между острым и хроническим лейкозом
Лейкоз — это рак лейкоцитов в организме; он развивается в костном мозге и лимфатической системе, а затем попадает в кровоток. Четыре типа лейкемии делятся на две категории: острые и хронические. Острый лейкоз поражает незрелые клетки, называемые стволовыми клетками, тогда как хронический лейкоз развивается в зрелых клетках. Однако об этих двух типах лейкемии, включая их подкатегории, нужно понять гораздо больше.
Острые лейкозы быстро прогрессируют, поскольку они поражают стволовые клетки, называемые бластами, которые быстро делятся — как нормальные клетки крови, так и раковые. Проблема в том, что аномальные клетки быстро проникают в кровь, потому что они не перестают делиться, когда должны. Поскольку они очень быстро растут, острые лейкозы часто распространяются на органы и центральную нервную систему человека. Многие люди с острыми формами лейкемии будут испытывать симптомы легкого кровотечения и синяков, усталости, непреднамеренной потери веса и частых инфекций.
Это серьезная форма рака. Без лечения прогноз острой лейкемии мрачный, большинство людей выживают всего несколько месяцев. Однако некоторые виды острого лейкоза можно вылечить при правильном лечении.
Существует два типа острого лейкоза:
Острый лимфоцитарный (также называемый лимфобластным) лейкоз (ОЛЛ): этот тип рака развивается, когда аномальные незрелые лимфоцитарные лейкоциты начинают бесконтрольно делиться.Зрелые лимфоциты ответственны за выработку антител.
Острый миелогенный (также называемый миелоидным) лейкоз (ОМЛ): это заболевание может начаться с незрелых белых кровяных телец, отличных от тех, которые становятся лимфоцитами. Он также развивается в других типах незрелых клеток крови — красных кровяных тельцах и тромбоцитах. ОМЛ имеет несколько подтипов, разделенных на категории по типу и зрелости клеток, в которых развивается заболевание.
У женщин вероятность развития ОЛЛ выше, чем у мужчин, особенно после 45 лет.У мужчин больше шансов заболеть всеми другими типами лейкозов. У детей также больше шансов заболеть ОЛЛ, чем ОМЛ. Около 75% всех детских лейкозов — это ВСЕ, остальные — ОМЛ.
Лечение может быть эффективным для людей с острым лейкозом, особенно с ОЛЛ. Более 80% людей имеют полную ремиссию болезни, а около 40% излечиваются. Типы лечения включают:
- Химиотерапия
- Трансплантация стволовых клеток, которая заменяет костный мозг человека новым костным мозгом
- Лучевая терапия, которая использует рентгеновские лучи для уничтожения раковых клеток, часто используется, когда рак распространился на центральную нервную систему. система
- Целевые препараты, которые атакуют определенный аспект раковых клеток
- Клинические испытания, проверяющие новые методы лечения и процедуры
При хроническом лейкозе аномальные клетки частично созрели и часто кажутся нормальными.Когда эти клетки развиваются в клетки лейкемии, они также не борются с болезнью и выживают дольше, чем нормальные лейкоциты, позволяя им накапливаться в крови.
Эти виды рака прогрессируют медленнее, чем острые лейкемии. Люди часто не проявляют никаких симптомов и могут жить много лет после развития болезни. Однако хронические лейкозы не поддаются лечению, что затрудняет их лечение.
Есть два типа хронического лейкоза:
Хронический лимфолейкоз (ХЛЛ): это заболевание развивается из частично созревших лейкоцитов, называемых лимфоцитами.Он начинается в костном мозге, но затем попадает в кровь. Есть два типа ХЛЛ: один растет медленно, а другой развивается быстрее и является более серьезным.
Хронический миелогенный лейкоз (ХМЛ): Эта форма лейкемии начинается с генетического изменения нелимфоцитарных белых кровяных телец, эритроцитов и тромбоцитов. Затем аномальный ген начинает превращать нормальные клетки в клетки ХМЛ. Хотя это медленнорастущий тип лейкемии, он также может перерасти в острый, быстрорастущий лейкоз.
У мужчин хронические лейкозы развиваются гораздо чаще, чем у женщин, хотя ХМЛ редко встречается у обоих полов и всех возрастных групп. Хронические лейкозы у детей встречаются редко.
Лечение включает:
- Целевые препараты
- Биологическая терапия моноклональными антителами: искусственные препараты, которые помогают иммунной системе убивать раковые клетки, в основном используются с CLL
- Биологическая терапия интерфероном: инъекция веществ, производимых в иммунной системе. Система, используемая в основном с ХМЛ
- Химиотерапия
- Трансплантация стволовых клеток
- Клинические испытания новых методов лечения и процедур
Понимание различий между острыми и хроническими формами лейкемии имеет решающее значение при формировании плана действий по борьбе с заболеванием.Поговорите с врачом о своих симптомах и о лучшем плане лечения для вашего состояния.
В чем разница между острым и хроническим лейкозом? Онкологический центр
Стволовые клетки происходят из нашего костного мозга и в процессе роста становятся эритроцитами, лейкоцитами и тромбоцитами. Эти клетки составляют нашу кровь и играют жизненно важную роль в нашем здоровье, доставляя кислород к нашим органам, помогая бороться с инфекциями и останавливая кровотечение в случае порезов.
Если эти клетки останавливаются в своем развитии, это может привести к лейкемии.В зависимости от типа поврежденных стволовых клеток и того, когда происходит нарушение их роста, лейкоз можно разделить на две группы: острые и хронические.
Наш Элиас Джаббур, доктор медицины, рассказывает о различиях между острыми и хроническими лейкозами, в том числе о том, как они делятся на подтипы и как это помогает в выборе лечения.
В чем разница между острым и хроническим лейкозом?
Как и все живые существа, клетки имеют жизненный цикл: они рождаются, созревают и затем умирают.У лейкемии этот жизненный цикл заблокирован. Детские клетки не созревают или взрослые клетки никогда не умирают. Лейкемии классифицируются в зависимости от того, когда клетки отклоняются от своего жизненного цикла.
- Хронический лейкоз возникает в результате сбоя в жизненном цикле после созревания клеток. Взрослые клетки часто в некоторой степени все еще могут функционировать, но, в конечном счете, они несовместимы с жизнью.
- Острый лейкоз возникает при остановке развития на ранних этапах жизненного цикла клетки.Эти детские клетки, находящиеся в тупике, не имеют никакой функции.
Как лимфоидные или миелоидные клетки подходят для диагностики лейкемии?
Помимо хронического и острого, диагноз лейкемии может быть дополнительно определен путем определения того, какой из двух типов стволовых клеток поражен: лимфоидные клетки или миелоидные клетки.
Лимфоидные клетки производят лимфоциты — белые кровяные тельца, составляющие большую часть иммунной системы. Миелоидные клетки производят эритроциты, тромбоциты и другие типы белых кровяных телец, не являющиеся лимфоцитами.
Хотя существуют более редкие формы болезни, большинство лейкозов делятся на четыре основных подтипа:
- острый миелоидный лейкоз
- острый лимфобластный лейкоз
- хронический миелолейкоз
- хронический лимфолейкоз
Независимо от того, когда в жизненном цикле происходит сбой и какие клетки поражены, раковые клетки вытесняют здоровые клетки и мешают им выполнять свою важную работу.
Различаются ли симптомы при хроническом и остром лейкозе?
Да, хронические лейкозы развиваются медленно, поэтому у большинства пациентов не так много симптомов.
Однако пациенты с острыми лейкозами обычно легко кровоточат, например, при чистке зубов. У них могут быть частые кровотечения из носа или обильные менструации. Также они часто испытывают жар и утомляемость.
Как лечат острый лейкоз?
Цель лечения — восстановить нормальное функционирование клеток. При остром лейкозе первый шаг называется индукцией. С помощью химиотерапии мы лечим раковые клетки крови, а также костный мозг.Чтобы убедиться, что все раковые клетки исчезли, существует второй этап лечения, называемый консолидацией. На этом этапе некоторые пациенты получают больше химиотерапии, а другие получают больше химиотерапии плюс трансплантацию стволовых клеток. Подход консолидации зависит от того, насколько агрессивен лейкоз, а также от уникальных генетических характеристик диагноза, который определяется с помощью серии тестов при первоначальном диагнозе.
Третий этап — это обслуживание, которое помогает снизить риск рецидива рака.В течение двух-трех лет пациенты принимают химиотерапию в основном в виде таблеток и попадают в больницу только раз в месяц. Мы использовали этот подход в течение некоторого времени при лечении острого лимфобластного лейкоза, но новое исследование показывает, что он помогает продлить жизнь и пациентам с острым миелоидным лейкозом.
Наконец, поскольку некоторые химиотерапевтические препараты не проходят через так называемый гематоэнцефалический барьер, мы использовали лучевую терапию для предотвращения рецидива. Но это может вызвать долгосрочные побочные эффекты, которые могут иметь большое влияние на наших педиатрических пациентов с лейкемией.Теперь мы используем интратекальную химиотерапию, которая вводится в позвоночник и распространяется до мозга.
Как лечат хронические лейкозы?
Любой диагноз рака — проблема, но хорошая новость заключается в том, что с хроническим лейкозом вы можете жить нормальной жизнью. Фактически, 40% пациентов с хроническим лимфолейкозом можно просто наблюдать и не нуждаться в лечении.
Когда приходит время лечить рак, мы используем таргетную терапию, поскольку пациенты с хроническими лейкозами, как правило, старше и имеют больше данных в истории болезни.
Химиотерапия убивает все в надежде, что раковые клетки умрут, а нормальные клетки восстановятся. Но поскольку он уничтожает все, наступает период, когда у пациента мало здоровых клеток крови, и он сталкивается с риском осложнений. С другой стороны, таргетная терапия атакует только раковые клетки, экономя все остальное и ограничивая риск осложнений. В настоящее время для лечения лейкозов используются несколько целевых препаратов по отдельности или в сочетании с химиотерапией, но их количество не за горами.
К сожалению, с хроническим миелоидным лейкозом нельзя наблюдать и ждать. Но лечение — это таблетки. Исторически сложилось так, что пациенты принимали таблетки всю оставшуюся жизнь, но новые исследования показывают, что если пациент чувствует себя хорошо через пять или шесть лет, мы можем прекратить химиотерапию с низким риском рецидива рака.
Хотите еще чем-нибудь рассказать о лечении лейкемии?
Я очень взволнован, потому что думаю, что мы вылечим лейкемию при моей жизни.Мы уже зашли так далеко. Например, до 2000 года выживаемость пациентов с острым лимфобластным лейкозом с положительной филадельфийской хромосомой составляла 10%. Таргетная терапия улучшила этот показатель до 35-40%, но пациенты должны были пройти трансплантацию стволовых клеток, что было непросто. Но я руководил исследованием препарата под названием понатиниб в сочетании с химиотерапией, и мы наблюдали, как выживаемость подскочила до 80% без трансплантата. Наша следующая цель — увеличить выживаемость до 90% и исключить необходимость в химиотерапии, используя комбинацию таргетной терапии и иммунотерапии.
Я горжусь тем, что доктор медицины Андерсон лидирует. Здесь мы разработали понатиниб, и теперь это международный стандарт. Мы являемся первопроходцами в лечении этих пациентов без химиотерапии.
Запишитесь на прием в MD Anderson онлайн или позвонив по телефону 1-877-632-6789.
Что такое лейкемия? | Комплексный онкологический центр Розуэлл-Парк
Лейкемия — это название, данное нескольким типам рака, которые начинаются в ткани, из которой образуются клетки крови.Чтобы понять лейкоз, полезно знать, как образуются нормальные клетки крови.
Roswell Park имеет специальные программы для пациентов с детской лейкемией, а также с хроническим лимфолейкозом (CLL). Посетите эти страницы, если вы ищете более подробную информацию.
Нормальные клетки крови
Большинство клеток крови развивается в костном мозге , мягком материале в центре большинства костей. Костный мозг содержит гематопоэтических стволовых клеток, , которые производят все виды клеток крови, в зависимости от того, какие типы клеток необходимы вашему организму в конкретное время.Эти стволовые клетки чрезвычайно заняты, производя миллиарды клеток крови каждый день. Когда любой из трех типов клеток крови стареет или повреждается, они умирают, а гемопоэтические стволовые клетки производят новые.
Каждому типу клеток крови посвящена особая работа:
- Лейкоциты , которые являются частью иммунной системы вашего организма, помогают бороться с болезнями. Есть несколько типов лейкоцитов.
- Красные кровяные тельца переносят кислород к тканям по всему телу.
- Тромбоциты помогают формировать сгустки крови, чтобы остановить кровотечение при травме.
Клетки лейкемии
Лейкемия — это просто еще одно название для рака крови . Когда у вас развивается лейкемия, ваш костный мозг начинает вырабатывать аномальные белые кровяные тельца, которые называются лейкозами и клетками. В отличие от нормальных клеток крови, лейкозные клетки не умирают, когда должны. Они могут вытеснять нормальные лейкоциты, эритроциты и тромбоциты.Это мешает нормальным клеткам крови выполнять свою работу — и именно тогда появляются симптомы.
Симптомы лейкемии
По мере роста лейкозных клеток в крови и костном мозге они вытесняют нормальные гемопоэтические стволовые клетки, которые затем не могут производить нормальное количество белых клеток (которые борются с инфекцией), красных кровяных телец (которые переносят кислород через ваше тело) и тромбоциты (которые помогают свертываться крови после травмы). Это мешает нормальным клеткам крови выполнять свою работу — и именно тогда появляются симптомы.У вас может развиться анемия (недостаток эритроцитов), лейкопения (недостаток лейкоцитов) или тромбоцитопения (недостаток тромбоцитов).
Узнайте о симптомах
Типы лейкемии
Существуют различные типы лейкемии, которые делятся на две основные группы — хронический и острый . Хронический лейкоз обычно ухудшается медленно, от месяцев до лет, в то время как острый лейкоз развивается быстро и прогрессирует в течение нескольких дней или недель.
Два основных типа лейкемии можно разделить на группы, основанные на типе пораженных лейкоцитов — лимфоид или миелоид .
Лимфоидные клетки являются частью иммунной системы. Они помогают бороться с инфекциями и контролировать рост рака. Лейкоз, который начинается в лимфоидных клетках, называется лимфоидным , лимфоцитарным или лимфобластным лейкозом .
Миелоидные клетки состоят из различных типов иммунных клеток, которые помогают бороться с бактериальными инфекциями; красные кровяные тельца, переносящие кислород по телу; и тромбоциты, которые свертывают кровь. Лейкоз, поражающий миелоидные клетки, называется миелоидным лейкозом , миелогенным или миелобластным лейкозом .
Хронический лейкоз
На начальных стадиях хронического лейкоза лейкозные клетки растут медленно, в течение периода от месяцев до лет, и все еще могут выполнять часть работы нормальных лейкоцитов, поэтому вы можете сначала не заметить никаких симптомов. Вот почему врачи часто обнаруживают хронический лейкоз с помощью лабораторных анализов, проводимых в рамках обычного осмотра.
Постепенно хронический лейкоз ухудшается. По мере увеличения количества лейкозных клеток в крови вы будете испытывать увеличение лимфатических узлов, увеличение органов, таких как печень и селезенка, в области желудка, а также инфекции или другие симптомы.Когда симптомы действительно появляются, они обычно сначала легкие, но постепенно ухудшаются.
- Хронический лимфолейкоз (ХЛЛ): ХЛЛ поражает лимфоидные клетки и обычно растет медленно. Ежегодно на его долю приходится около 21 000 новых случаев лейкемии. Большинство людей, у которых диагностировано это заболевание, старше 55 лет. Оно почти никогда не поражает детей. Поскольку ХЛЛ может быть похож на другие виды рака лимфатического узла (называемые лимфомами , ), в Розуэлл-Парке ХЛЛ обычно лечат врачи из группы лимфомы.Посетите эту страницу для получения дополнительной информации о CLL.
- Хронический миелоидный лейкоз (ХМЛ): ХМЛ поражает все типы миелоидных клеток и сначала обычно растет медленно. Обычно у пациентов обнаруживается повышенное количество лейкоцитов и генетическая аномалия, называемая Филадельфийская хромосома . Чаще всего ХМЛ лечат специальными химиотерапевтическими таблетками. Ежегодно на его долю приходится около 8 430 новых случаев лейкемии. Он поражает в основном взрослых в возрасте от 40 до 50 лет.
Джеймс Томпсон, доктор медицины, врач группы по лечению лейкемии в Розуэлл-парке, играет национальную роль в установлении стандартов лечения ХМЛ в качестве члена Группы рекомендаций по ХМЛ Национальной комплексной онкологической сети (NCCN) . Руководящие принципы NCCN определяют наилучшие способы предотвращения, обнаружения и лечения различных типов рака и являются наиболее широко используемыми стандартами в Соединенных Штатах.
Острый лейкоз
При остром лейкозе лейкозные клетки не могут выполнять какую-либо работу нормальных белых кровяных телец.Количество лейкозных клеток быстро увеличивается, поэтому болезнь быстро ухудшается.
Многие пациенты с острым лейкозом имеют симптомы или анализ крови, требующие срочного обследования и диагностики. Если у вас есть этот диагноз и вы хотите, чтобы вас осмотрели как можно скорее, позвоните по телефону 1-800-ROSWELL (1-800-767-9355).
- Острый миелоидный лейкоз (AML): AML — наиболее распространенный тип агрессивного лейкоза у взрослых. Этот рак включает быстрый рост аномальных миелоидных клеток и очень быстро растет, что приводит к очень высокому количеству белых кровяных телец или очень низкому количеству белых кровяных телец.Ежегодно на его долю приходится более 19 500 новых случаев лейкемии. Встречается в основном у взрослых.
- Острый лимфоцитарный (лимфобластный) лейкоз (ОЛЛ): ОЛЛ является наиболее распространенным типом рака. Это агрессивный рак крови, чаще всего встречающийся у младенцев, детей и молодых людей. Пациентов младше 18 лет обычно лечат врачи Розуэлл-Парка в нашей группе детской гематологии и онкологии. Этот рак, который также может поражать взрослых, является причиной почти 6000 новых случаев лейкемии каждый год.
Юнис С. Ван, доктор медицинских наук, руководитель службы по лечению лейкемии в Розуэлл-парке, играет на национальном уровне роль в установлении стандартов лечения как ОМЛ, так и ОЛЛ в качестве члена групп рекомендаций по ОМЛ и ВСЕЙ национальной общенациональной комиссии по лечению рака. Сеть (NCCN). Рекомендации NCCN определяют лучшие способы профилактики, выявления и лечения различных типов рака и являются наиболее широко используемыми стандартами в США
.Кара Келли, доктор медицинских наук, заведующая кафедрой детской онкологии, играет национальную роль в установлении стандартов лечения педиатрического ОЛЛ в качестве члена группы рекомендаций Национальной комплексной онкологической сети (NCCN) по лечению острого лимфобластного лейкоза у детей.
Редкие лейкемии
Несколько других, более редких типов лейкемии вызывают более 6000 новых диагнозов лейкемии каждый год. К ним относятся неоплазма бластных плазмоцитоидных дендритных клеток (BPDCN) и волосатоклеточный лейкоз.
Бластное плазмоцитоидное новообразование дендритных клеток
Этот рак, также известный как BPDCN, представляет собой очень редкий рак крови, который чаще всего проявляется кожными узелками или аномалиями, а также аномальными показателями крови и иногда опухолью в различных лимфатических узлах по всему телу.Диагноз может быть поставлен после биопсии кожи или с использованием образцов крови или костного мозга и может потребовать специализированного патологического исследования.
Таграксофусп-эраз (Эльзонрис ™), новый препарат для лечения этого рака, был одобрен FDA в декабре 2018 года. Розуэлл-Парк был одним из восьми центров по всей стране, предлагающих клинические испытания Эльзонриса, предоставляя нашим пациентам доступ к препарату. до утверждения FDA. Служба лейкемии Розуэл-Парка предлагает клинические испытания других многообещающих новых методов лечения как для недавно диагностированной, так и для устойчивой к лечению BPDCN.
Волосатоклеточный лейкоз
Волосатоклеточный лейкоз возникает, когда костный мозг начинает производить аномальных лимфоцитов или лейкозных клеток, которые под микроскопом выглядят волосатыми.
Волосатоклеточный лейкоз чаще диагностируется у мужчин старшего возраста. Поскольку эти клетки могут расти медленно или вообще не расти, лечение может не потребоваться. Многие пациенты проходят активное наблюдение — также называемое бдительным ожиданием — для наблюдения за ситуацией.Лечение можно начинать всякий раз, когда болезнь начинает ухудшаться. Он может включать химиотерапию, таргетную терапию, хирургическое вмешательство или биологическую терапию. Хотя волосатоклеточный лейкоз нельзя вылечить, лечение может привести к ремиссии на многие годы, поэтому это заболевание считается хроническим.
Записаться на прием
Узнать о леченииЧем ХЛЛ отличается от других типов лейкемии
Хронический лимфолейкоз (ХЛЛ) начинается в костном мозге, мягкой внутренней части ваших костей.Есть некоторые ключевые различия, которые отличают ХЛЛ от других типов лейкемии, таких как ОЛЛ (острый лимфолейкоз), ОМЛ (острый миелоидный лейкоз) и ХМЛ (хронический миелоидный лейкоз).
Кто заболеет
ХЛЛ — наиболее распространенная лейкемия у взрослых. Большинству людей диагноз ставят в возрасте от 60 до 70 лет. С возрастом ваш риск увеличивается. Другие типы, такие как ОЛЛ и ОМЛ, в основном возникают в детстве. Дети обычно не болеют медленнорастущими формами лейкемии.
Большинство людей живут с ХЛЛ намного дольше, чем с другими видами рака или другими видами лейкемии.
Где это начинается
ХЛЛ и ВСЕ начинаются в костном мозге, где обычно образуются клетки крови. В частности, он начинается в клетке костного мозга, называемой лимфоцитами. (Это то, что обозначает первая буква «L» в словах CLL и ALL.)
Лимфоциты становятся разновидностью лейкоцитов, которые помогают вашему организму бороться с инфекциями. Но при ХЛЛ лимфоциты бесконтрольно разрастаются. Анализы крови часто показывают высокий уровень лейкоцитов или лимфоцитов. Вам нужно будет пройти еще несколько анализов, чтобы узнать, лейкемия ли это.
Два других типа лейкемии — острый миелоидный лейкоз (ОМЛ) и хронический миелоидный лейкоз (ХМЛ) — начинаются в миелоидных клетках костного мозга. Миелоидные клетки развиваются в белые кровяные тельца, отличные от лимфоцитов, красные кровяные тельца (которые переносят кислород) и тромбоциты (которые образуют сгустки, помогающие остановить кровотечение).
Как быстро растут раковые клетки
Буква «C» в CLL означает хронический. Это означает, что рак обычно растет и распространяется медленно, хотя ХЛЛ также может расти быстрее.
Напротив, типы лейкозов, в названии которых есть слово «острый» (острый лимфоцитарный лейкоз и острый миелоидный лейкоз), растут намного быстрее, чем большинство хронических лейкозов. Люди с этим типом обычно очень больны и нуждаются в немедленном лечении.
Если, как и у большинства людей с ХЛЛ, у вас медленнорастущий тип, вы можете не замечать никаких симптомов в течение многих лет. Обычный анализ крови может быть первым признаком того, что что-то не так. Вам может не понадобиться лечение сразу. (Ваш врач, конечно, будет внимательно следить за вашим здоровьем.) Нужно ли вам начинать лечение, зависит от ваших симптомов, результатов анализов и стадии (от 0 до IV) вашего заболевания.
Врачи могут проверять общие и необычные белки на поверхности клеток ХЛЛ. Результаты помогают диагностировать и лечить его, а также проверять прогноз (как это состояние может повлиять на вас).
Хронический лейкоз в сравнении с острым: понимание различий
Лейкемии — это злокачественные опухоли, которые развиваются из клеток крови. Хотя существует несколько различных типов лейкемии, их обычно классифицируют как «хронические» или «острые».«Основное различие между этими двумя категориями заключается в том, что хронические лейкозы растут медленно, а острые лейкозы — быстро. Поскольку острый и хронический рак прогрессируют с разной скоростью, они могут вызывать разные наборы симптомов и по-разному реагировать на лечение.
Нормальная функция клеток кровиПонимание клеток крови и их развития может пролить свет на разницу между хроническим и острым лейкозом.
Ваша кровь состоит из нескольких различных типов клеток, включая эритроциты, лейкоциты и тромбоциты.Каждая из этих клеток играет разную роль в поддержании здоровья вашего тела.
- Красные кровяные тельца доставляют кислород клеткам по всему телу и помогают избавиться от продуктов жизнедеятельности.
- Лейкоциты борются с инфекциями, атакуя и удаляя инородные частицы, такие как микробы.
- Тромбоциты образуют сгустки крови и помогают предотвратить слишком сильное кровотечение.
Все эти клетки крови развиваются из незрелых клеток, называемых гемопоэтическими стволовыми клетками (ГСК). HSC живут в костном мозге, мягких тканях, которые заполняют внутреннюю часть костей.Чтобы создать новые клетки крови, HSC делают копии самих себя. Некоторые из копий представляют собой немного более зрелые клетки, называемые клетками-предшественниками. Клетки-предшественники растут и делятся, чтобы произвести другие клетки, которые стали еще более зрелыми. Этот процесс повторяется до тех пор, пока не сформируются полностью зрелые клетки крови. HSC ежедневно производят миллиарды новых клеток крови.
Что такое лейкемия?Клетки содержат гены, которые действуют как инструкции, определяющие работу, которую клетка выполняет в организме.Когда гены претерпевают определенные изменения, может развиться рак. Эти изменения генов могут затруднить восстановление повреждений клеткой или могут ускорить рост клетки. Некоторые люди рождаются с этими злокачественными изменениями генов, в то время как у других они развиваются позже, когда клетки повреждаются.
Лейкемия возникает, когда в одной клетке крови развиваются определенные изменения генов, вызывающих рак. Лейкемия обычно начинается в незрелой стволовой клетке или клетке-предшественнике. Если одна из этих клеток повреждается, она может стать злокачественной и выйти из-под контроля.Это может вызвать несколько больших проблем:
- Раковые клетки ненормальные и незрелые и не могут выполнять функции нормальных клеток крови.
- Раковые клетки быстро размножаются и захватывают костный мозг и кровь, вытесняя нормальные клетки крови.
- Костный мозг больше не будет производить столько нормальных кровяных телец, как раньше, что приводит к проблемам по всему телу.
Есть несколько типов лейкемии.Эксперты классифицируют лейкоз как миелоидный или лимфоидный в зависимости от того, какой тип клеток стал злокачественным. Миелоидные клетки производят тромбоциты, эритроциты и некоторые типы белых кровяных телец, а лимфоидные клетки образуют другие типы белых кровяных телец, называемые лимфоцитами. Кроме того, эксперты классифицируют эти типы лейкемии как острые или хронические в зависимости от скорости их роста.
Острый лейкозОстрый лейкоз развивается и очень быстро ухудшается. Клетки острого лейкоза очень незрелые и не могут выполнять ни одну из задач, которые должны выполнять клетки крови.Если у вас острый лейкоз, вы, скорее всего, почувствуете себя плохо в течение нескольких недель после развития болезни. Вам нужно будет немедленно начать лечение.
Хронический лейкозХронический лейкоз растет медленнее. Может пройти несколько месяцев или даже лет, прежде чем вы начнете испытывать симптомы. Клетки хронического лейкоза более зрелые, чем клетки острого лейкоза, но они еще не полностью развиты. Они могут выполнять некоторые функции нормальных кровяных телец, но они все равно не работают должным образом.
Основные типы лейкозовНа основании приведенных выше классификаций существует четыре основных типа лейкозов:
- Острый миелоидный лейкоз — это наиболее распространенный тип острого лейкоза у взрослых.
- Острый лимфоцитарный лейкоз — чаще всего этого типа является лейкоз у детей.
- Хронический миелоидный лейкоз — эта форма лейкемии чаще встречается у взрослых, чем у детей.
- Хронический лимфолейкоз — это наиболее распространенный тип хронического лейкоза, наблюдаемый у взрослых.
Каждый из этих четырех типов лейкемии также состоит из множества подтипов. Существуют и другие типы лейкемии, но они часто встречаются редко. Узнайте больше о видах лейкемии.
Симптомы острого лейкоза по сравнению с хроническимПоскольку острые и хронические лейкозы прогрессируют с разной скоростью, они могут выглядеть и ощущаться по-разному в организме.
Острый лейкоз с большей вероятностью вызывает тяжелые симптомы, которые проявляются на ранней стадии заболевания. Симптомы острого лейкоза могут включать:
- Усталость
- Одышка
- Бледная кожа
- Синяки или обесцвеченные пятна под кожей
- Проблемы с кровотечением
- Неожиданная потеря веса
- Боль в костях или суставах
- Лихорадка
- Часто Инфекции
Симптомы хронического лейкоза обычно менее выражены и могут появиться только на более поздних стадиях заболевания.Симптомы хронического лейкоза включают:
- Чувство усталости или слабости
- Увеличение лимфатических узлов, которое проявляется в виде шишек в подмышечных впадинах, шее или паху
- Боль в животе и вздутие живота
- Чувство переполнения желудка
- Лихорадка или ночная потливость
Чаще всего симптомы, обнаруживаемые как при остром, так и при хроническом лейкозе, вызваны другими заболеваниями. Однако, если вы испытываете какие-либо из этих симптомов, поговорите со своим врачом.
Диагностика различных типов рака кровиУ детей чаще диагностируется острый лейкоз, тогда как хронические лейкемии чаще диагностируются у взрослых. Врачи могут определить, какой тип клеток стал злокачественным, с помощью различных диагностических тестов, включая общий анализ крови, мазок периферической крови, проточную цитометрию и генетические тесты. Узнайте больше о тестах, используемых для диагностики лейкемии.
Сравнение лечения острого лейкоза и хронического лейкозаОстрые лейкозы быстро прогрессируют, но их часто легче вылечить, чем хронические лейкозы.Хотя лечение хронических лейкозов может не сработать, хронические раковые опухоли медленно растут и медленно вызывают проблемы. Часто люди могут долго жить с хроническим миелоидным лейкозом или хроническим лимфолейкозом.
Для лечения лейкемии можно использовать многие виды терапии. Рекомендуя план лечения, ваш врач примет во внимание множество факторов, в том числе ваш возраст, другие проблемы со здоровьем, которые могут у вас возникнуть, распространился ли лейкоз и ваш тип лейкемии.
Лечение лейкемии может включать следующее:
- Химиотерапия — химиотерапевтические препараты, которые могут убивать быстрорастущие раковые клетки, можно назначать отдельно или в сочетании с другими лекарствами.
- Таргетная терапия — Таргетная терапия может распознавать и атаковать определенные молекулы, обнаруженные только в лейкозных клетках.
- Иммунотерапия — процедуры иммунотерапии укрепляют вашу иммунную систему и позволяют ей бороться с лейкемией.
- Трансплантация стволовых клеток — Во время трансплантации стволовых клеток ваши клетки костного мозга погибают, а затем вы получаете новые HSC, которые могут создавать новые доброкачественные клетки крови.
- Лучевая терапия — Лучевая терапия использует лучи энергии, такие как рентгеновские лучи, для повреждения раковых клеток.
- Клинические испытания — В ходе клинических испытаний исследователи проверяют, являются ли методы лечения рака безопасными и эффективными. Спросите своего врача, могут ли клинические испытания быть для вас хорошим вариантом, поскольку они предоставляют доступ к новым методам лечения, которые еще не входят в стандартную помощь.
Навигация по новому диагнозу может быть сложной задачей, но наличие сети поддерживающих людей, которые понимают, может помочь облегчить эту задачу.MyLeukemiaTeam — это социальная сеть для людей, больных лейкемией, и их близких. Присоединяясь, вы получаете группу поддержки из более чем 7500 человек. Участники понимают, каково жить с лейкемией, и готовы дать совет, поддержку и ответы, когда вы попытаетесь узнать больше об этой болезни.
Был ли у вас или вашего близкого недавно диагностирован острый или хронический лейкоз? Прокомментируйте свой опыт ниже или опубликуйте сообщение в MyLeukemiaTeam, чтобы начать разговор.
типов | Хронический миелоидный лейкоз
Выделяют 2 основные группы лейкозов — острый и хронический лейкоз.Затем они группируются по типу пораженных лейкоцитов.
В зависимости от того, является ли лейкоз острым или хроническим, определяется, насколько быстро лейкоз может развиться и ухудшиться.
Острый и хронический лейкоз
Острые лейкемии, как правило, быстро развиваются и быстро ухудшаются, если их не лечить. Хронические лейкемии развиваются медленно и имеют тенденцию к ухудшению медленно, в течение длительного времени.
При хроническом лейкозе белые кровяные тельца почти полностью развиты, но не являются полностью нормальными.Они по-прежнему работают, но не так хорошо, как должны, в борьбе с инфекцией. В вашем организме вырабатывается слишком много аномальных белых кровяных телец.
Типы хронических лейкозов
CML и CLL
Двумя наиболее распространенными типами хронического лейкоза являются:
- хронический миелоидный лейкоз (ХМЛ)
- хронический лимфолейкоз (ХЛЛ)
Разница между ними — тип лейкоцита, ставшего злокачественным.При ХЛЛ аномальные клетки развиваются из ранних клеток крови, называемых лимфоидными стволовыми клетками крови. Раковые лейкоциты — это В-лимфоциты, также называемые В-клетками.
При ХМЛ аномальные лейкозные клетки развиваются из ранних клеток крови, называемых миелоидными стволовыми клетками крови. Они становятся миелоцитами. Эти клетки иногда называют гранулоцитами. Таким образом, вы можете услышать этот тип лейкемии, называемый хроническим гранулоцитарным лейкозом или ХГЛ.
Волосатоклеточный лейкоз
Существует третий тип хронического лейкоза, называемый волосатоклеточным лейкозом.Это реже, чем CLL или CML. Клетки лейкемии имеют кусочки, которые выступают из поверхности клетки и выглядят как волосы. Их можно увидеть под микроскопом, что и дало название этому типу лейкемии.
Хронический лимфолейкоз | CLL
Что такое лейкемия?
Лейкоз — это термин, обозначающий рак клеток крови. Лейкемия начинается в кроветворных тканях, таких как костный мозг. Костный мозг вырабатывает клетки, которые превращаются в лейкоциты, эритроциты и тромбоциты.У каждого типа ячейки своя работа:
- Белые кровяные тельца помогают организму бороться с инфекциями
- Красные кровяные тельца доставляют кислород из легких к тканям и органам
- Тромбоциты способствуют образованию тромбов, останавливающих кровотечение
Когда у вас лейкемия, ваш костный мозг производит большое количество аномальных клеток. Эта проблема чаще всего возникает с лейкоцитами. Эти аномальные клетки накапливаются в костном мозге и крови. Они вытесняют здоровые клетки крови и мешают вашим клеткам и крови выполнять свою работу.
Что такое хронический лимфолейкоз (ХЛЛ)?
Хронический лимфолейкоз (ХЛЛ) — это тип хронического лейкоза. «Хронический» означает, что лейкоз обычно ухудшается медленно. При ХЛЛ костный мозг производит аномальные лимфоциты (тип лейкоцитов). Когда аномальные клетки вытесняют здоровые, это может привести к инфекции, анемии и легкому кровотечению. Аномальные клетки также могут распространяться вне крови в другие части тела. ХЛЛ — один из наиболее распространенных типов лейкемии у взрослых.Часто возникает в среднем возрасте или после него. У детей встречается редко.
Что вызывает хронический лимфолейкоз (ХЛЛ)?
ХЛЛ возникает при изменении генетического материала (ДНК) в клетках костного мозга. Причина этих генетических изменений неизвестна, поэтому трудно предсказать, кто может заразиться ХЛЛ. Есть несколько факторов, которые могут повысить ваш риск.
Кто подвержен риску хронического лимфолейкоза (ХЛЛ)?
Трудно предсказать, кто заболеет ХЛЛ. Есть несколько факторов, которые могут повысить ваш риск:
- Возраст — риск возрастает с возрастом.Большинство людей с диагнозом ХЛЛ старше 50 лет.
- Семейный анамнез ХЛЛ и других заболеваний крови и костного мозга
- Расовая / этническая группа — ХЛЛ чаще встречается у белых, чем у людей из других расовых или этнических групп
- Воздействие определенных химикатов, в том числе Agent Orange, химического вещества, которое использовалось во время войны во Вьетнаме.
Каковы симптомы хронического лимфолейкоза (ХЛЛ)?
Вначале ХЛЛ не вызывает никаких симптомов.Позже у вас могут появиться такие симптомы, как
- Увеличение лимфатических узлов — вы можете заметить их как безболезненные шишки на шее, подмышках, животе или паху
- Слабость или чувство усталости
- Боль или чувство распирания под ребрами
- Лихорадка и инфекция
- Легкие синяки или кровотечения
- Петехии — крошечные красные точки под кожей. Они вызваны кровотечением.
- Потеря веса по неизвестной причине
- Обливание ночного пота
Как диагностируется хронический лимфолейкоз (ХЛЛ)?
Ваш лечащий врач может использовать множество инструментов для диагностики ХЛЛ:
- Медицинский осмотр
- История болезни
- Анализы крови, такие как полный анализ крови (CBC) с дифференциальными и биохимическими тестами.В биохимических анализах крови измеряются различные вещества в крови, включая электролиты, жиры, белки, глюкозу (сахар) и ферменты. Специфические биохимические тесты крови включают базовую метаболическую панель (BMP), комплексную метаболическую панель (CMP), функциональные тесты почек, функциональные тесты печени и панель электролитов.
- Тесты проточной цитометрии, которые проверяют наличие лейкозных клеток и определяют, какой это тип лейкемии. Тесты могут проводиться на крови, костном мозге или других тканях.
- Генетические тесты для поиска изменений генов и хромосом
Если вам поставили диагноз ХЛЛ, вам могут потребоваться дополнительные тесты, чтобы определить, распространился ли рак.К ним относятся визуализационные тесты и тесты костного мозга.
Какие методы лечения хронического лимфолейкоза (ХЛЛ)?
Лечение ХЛЛ включает
- Бдительное ожидание, что означает, что вы не получите лечение сразу. Ваш лечащий врач регулярно проверяет, появляются ли или изменяются ваши признаки или симптомы.
- Таргетная терапия, при которой используются лекарства или другие вещества, которые атакуют определенные раковые клетки с меньшим вредом для нормальных клеток.