Мышечная дистрофия | Неврология | Заболевания
Мышечная дистрофия – это патологическое заболевание, которое характерно для людей, ведущих лежачий образ жизни. Также данное заболевание может появится у людей с острыми хроническими заболеваниями мышц и костей.
Виды
Мышечная дистрофия очень распространенное патологическое заболевание. Бывает детская и взрослая дистрофия мышц. Также мышечная дистрофия имеет наследственный характер (генетическая и наследственная дистрофия). По характеру и месту локализации различают:
-
инфекционную и неинфекционную;
-
миотоническую;
-
тазово-плечевую;
-
врожденную;
-
плечелопаточную;
.
Симптомы
Мышечная дистрофия прогрессирует заболевание мышечной слабости и потери трудоспособности
Диагностика
Обратиться к доктору следует немедленно, как только вы заметили мышечную слабость.. К методам диагностики на данном этапе развития медицины относят МРТ. Оно покажет анатомические и физиологические изменения в организме. При этом заболевании также стоит сдать общий анализ крови, мочи и кала. После проведенных диагностик доктор поставит диагноз и направит на лечение.
Лечение
Лечение проводиться с помощью комплексной терапии: консервативное лечение и физиотерапия. Еще не разработано лечение, которые бы полностью устранило это заболевание.
Профилактика
Лечение мышечной дистрофии продолжается его профилактикой. Очень важно после выписки с госпиталя не забыть о приписках врача. Пациент должен снизить к минимуму физические нагрузки, стрессовые ситуации, вести здоровый образ жизни, отказаться от алкоголя, наркотиков и курения.
Прогрессирующие мышечные дистрофии | МКДЦ ФГБНУ НЦН
Прогрессирующие мышечные дистрофии (ПМД) — гетерогенная группа наследственных заболеваний, характеризующихся прогрессирующей мышечной слабостью и атрофией скелетных мышц.
КЛИНИЧЕСКАЯ КАРТИНА
Для всех ПМД типичны мышечная слабость различной степени выраженности и мышечные атрофии. Тип распределения мышечной слабости при ПМД — один из основных диагностических критериев. Для каждой из форм ПМД характерно избирательное поражение определённых мышц при сохранности других, рядом расположенных. В целом типичный миопатический симптомокомплекс включает следующие признаки.
• Симметричную проксимальную мышечную слабость различной степени выраженности (мышечная сила от 3-4 баллов на ранней и до 1-0 — на поздних стадиях заболевания) , постепенно развивающиеся атрофии мышц.
• Симптом Говерса: больной, для того чтобы подняться из положения на корточках, опирается руками об пол, затем поднимается, опираясь руками об колени, — «взбирается по себе». Этот рано появляющийся симптом обусловлен слабостью мышц бёдер и тазового пояса.
• Затруднения при ходьбе по лестнице — больной помогает себе с помощью рук.
• «Утиную» (переваливающуюся) походку, связанную со слабостью мышц тазового пояса.
• Поясничный гиперлордоз, обусловленный слабостью мышц тазового пояса и спины.
• «Крыловидные» лопатки вследствие слабости передней зубчатой мышцы, а также других мышц, фиксирующих лопатку.
• Псевдогипертрофию икроножных мышц вследствие развития в них соединительной ткани (сила мышц при этом снижена) .
• Ходьбу на цыпочках из-за контрактур ахилловых сухожилий.
• Сохранность экстраокулярных мышц, мышц лица.
Миопатический симптомокоплекс наиболее отчётливо выявляют при ПМД Дюшенна и Беккера.
• Для ПМД Дюшенна характерно раннее начало заболевания (в 3-7 лет) , быстрое прогрессирование, высокие показатели КФК, выраженная спонтанная активность по данным игольчатой ЭМГ, отсутствие дистрофина в мышцах при иммуногистохимическом исследовании. По мере прогрессирования мышечной слабости затрудняется самостоятельная ходьба и уже в 9-15 лет больные вынуждены пользоваться инвалидным креслом, что провоцирует развитие кифосколиоза, остеопороза. На поздних стадиях у большинства больных развиваются дилатационная кардиомиопатия, слабость дыхательной мускулатуры. Интеллект чаще всего умеренно снижен.
• Клинические про явления ПМД Беккера в целом напоминают таковые при форме Дюшенна, но течение заболевания более мягкое: дебют приходится на более поздний возраст (от 2 до 21 года, в среднем в 11 лет) , летальный исход наступает позже (в 23-63 года) .
• Конечностно-поясные формы ПМД также характеризуются развитием миопатического симптомокомплекса. ПМД Эрба по возрасту начала заболевания, скорости прогрессирования и клиническим проявлениям напоминает форму Беккера, однако для неё не характерна кардиальная патология, кроме того, заболевание отмечают как у мальчиков, так и девочек. При других конечностнопоясных формах возможны слабость мышц лица и кардиомиопатия.
• ПМД Ландузи-Дежерина характеризуется выраженной слабостью мимических мышц (за исключением редкой формы без мимической слабости), симптомом «крыловидных» лопаток, слабостью дву- и трёхглавых мышц плеча при интактных дельтовидных мышцах, степпажем. Как правило, интактными остаются экстраокулярные мышцы (за исключением одного подтипа) и мышцы языка и глотки, дыхательная мускулатура. У некоторых больных возникает слабость мышц тазового пояса (около 20% больных вынуждены пользоваться инвалидным креслом) . Мышечные атрофии часто бывают асимметричными. У многих больных отмечают снижение слуха, кардиомиопатию или нарушения сердечного ритма.
У многих больных отмечают снижение слуха, кардиомиопатию или нарушения сердечного ритма.
• ПМД Эмери-Дрейфуса характеризуется наличием контрактур (чаще в локтевых, коленных суставах, задних мышцах шеи, из-за которых голова оказывается слегка запрокинутой) и плечелопаточно-перонеальным распределением мышечной слабости и атрофий с сохранностью лицевой мускулатуры. Часто отмечают нарушения ритма сердца и кардиомиопатию. Заболевание часто дебютирует с контрактур.
• Основной симптом офтальмофарингеальной формы — хроническая прогрессирующая наружная офтальмоплегия, затем присоединяется умеренный бульбарный синдром. В дальнейшем развивается проксимальная мышечная слабость в руках и ногах.
• Дистальные миопатии характеризуются преобладанием слабости дистальных мышц. При миопатии Веландер в наибольшей степени поражаются разгибатели кистей, при миопатии Миоши — икроножные мышцы: больные плохо стоят на носках, часто спотыкаются. При миопатии Говерса, тибиальной миопатии главный симптом — степпаж из-за слабости перонеальной группы мышц, при этом миопатия Говерса склонна к дальнейшей генерализации: через 5-10 лет присоединяется слабость кистей и мышц шеи, часто отмечают «свисание» 1 пальца на ногах и V — на руках. При тибиальной миопатии, распространённой в Финляндии, чаще всего наблюдают изолированное поражение передних больше берцовых мышц, иногда развивается кардиомиопатия.
СИМПТОМЫ
При ПМД Дюшенна, Беккера, конечностно-поясных формах проявляется наиболее выраженная слабость в пояснично-подвздошных мышцах, мышцах бёдер, дельтовидных, дву- и трёхглавых мышцах плеча. Менее выражена слабость в дистальных мышцах конечностей. Лицевые мышцы остаются сохранными. Наряду с мышечной слабостью постепенно развиваются гипотрофии поражённых мышц вплоть до атрофии на поздних стадиях. При этом соседние мышцы могут быть полностью клинически интактны.
ЛЕЧЕНИЕ
Лечение должно производиться исключительно врачом-неврологом. Самолечение недопустимо. В настоящий момент радикального лечения ПМД не существует. Цель лечения — поддержание мышечной силы, предупреждение развития контрактур, деформаций суставов.
В настоящий момент радикального лечения ПМД не существует. Цель лечения — поддержание мышечной силы, предупреждение развития контрактур, деформаций суставов.
Немедикаментозное лечение
Чрезмерная физическая нагрузка, как и недостаточная, приводит к нарастанию мышечной слабости. Ежедневная ЛФК позволяет поддерживать мышечный тонус и препятствует развитию контрактур. Комплекс ЛФК обязательно должен включать активные и пассивные упражнения, упражнения на растяжку/предупреждение контрактур и дыхательную гимнастику. Активный массаж с разминанием мышц может усиливать мышечную слабость и утомляемость, поэтому рекомендуют щадящий массаж. Физиотерапевтическое лечение больные переносят по-разному: некоторые не ощущают улучшений или даже жалуются на усиление мышечной слабости.
Хирургическое лечение
В некоторых случаях возможно хирургическое лечение контрактур, однако при этом необходимо помнить о возможности увеличения мышечной слабости за время восстановительного лечения (вплоть до потери способности к ходьбе). В ряде случаев необходима имплантация кардиостимулятора.
Дистрофия
Это сложный патологический процесс, в основе которого лежат нарушения клеточного метаболизма, приводящие к структурным изменениям. При дистрофии происходит повреждение клеток и межклеточного вещества, что влечет за собой изменение функций органа. Патологический процесс обусловлен нарушением трофики — совокупности механизмов, которые отвечают за сохранность структуры клеток и тканей и обеспечивают метаболизм.
Причины дистрофии
В основе дистрофических процессов лежат мутации аутосомно-доминантного генома, основная функция которого состоит в синтезировании и регенерации в теле человека специфического белка, необходимого для полноценного формирования мышечных волокон.
Формы патологии
По виду обменных нарушений дистрофия делится на белковую, углеводную, жировую, минеральную; по распространенности — на местную и общую; по локализации — на клеточную, внеклеточную, смешанную; по этиологии — на врожденную и приобретенную.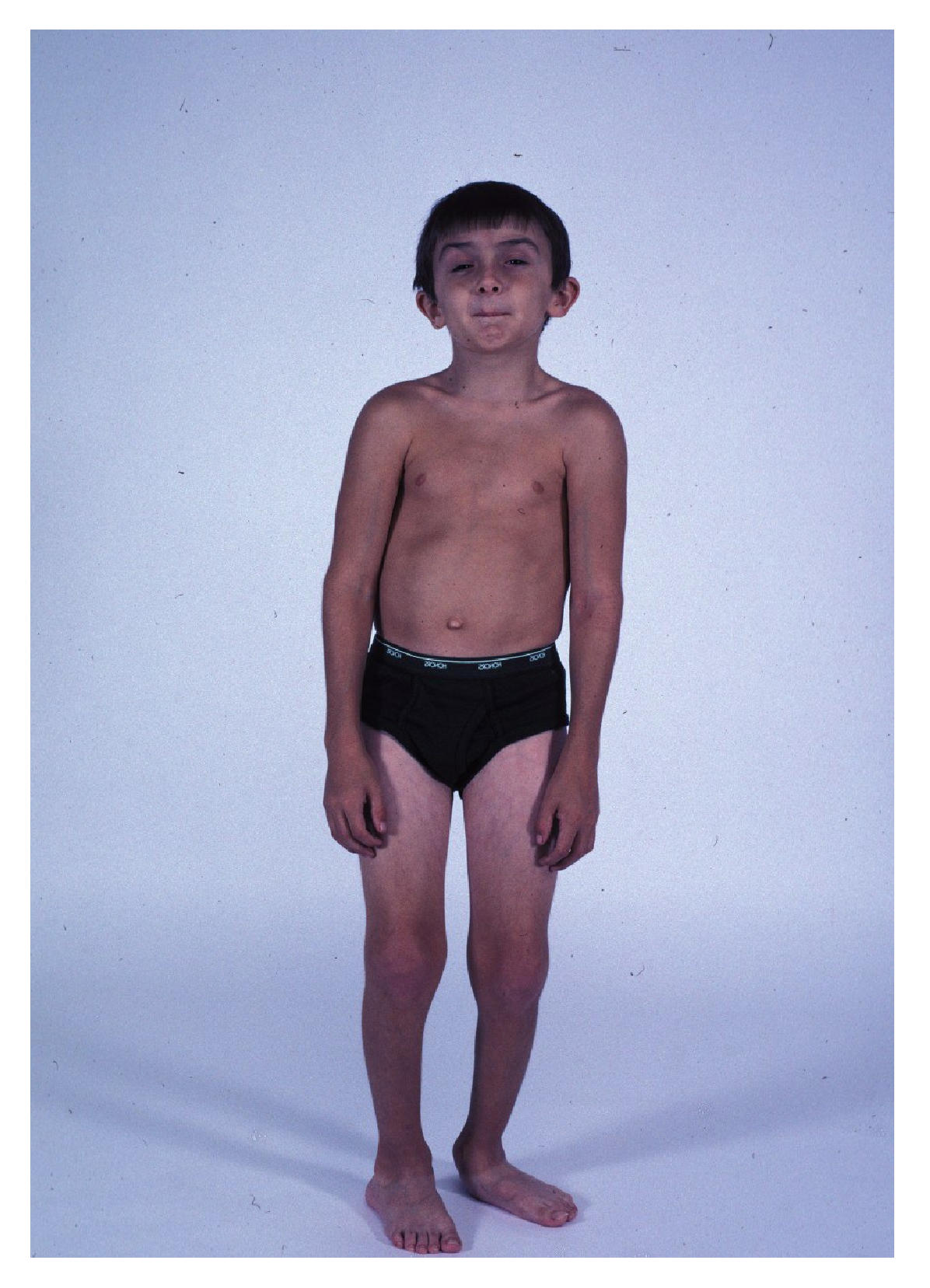 Миодистрофия — группа хронических заболеваний мышечных структур (в основном скелетных мышц).
Миодистрофия — группа хронических заболеваний мышечных структур (в основном скелетных мышц).
Врожденные формы мышечной дистрофии отличаются аутосомно-доминантным типом наследования. При этом в организме практически полностью отсутствует дистрофин — белок, необходимый для поддержания мышечной структуры. При некоторых формах патологии он вырабатывается в достаточном количестве, но неправильно функционирует.
Прогрессирующая мышечная дистрофия является эссенциально прогрессирующей дегенерацией мышечной ткани, которая развивается без поражения какого-либо отдела нервной системы и приводит к тяжелым атрофиям, слабости мышц определенной группы. Такие заболевания являются генетически обусловленными.
Прогрессирующие мышечные дистрофии Дюшенна и Беккера — Х-сцепленные рецессивные заболевания, которые развиваются из-за дефицита дистрофина. Его выработку кодирует ген DMD (маркер Xp21.2).
Прогрессирующая мышечная дистрофия Эмери – Дрейфуса ассоциирована с несколькими каузативными генами, кодирующими убиквитарные белки. Это ген EMD в локусе Xq28, ген FHL1 в локусе Xq26.3, ген LMNA в локусе 1q21.2, ген TMEM43.
Прогрессирующая мышечная дистрофия Ландузи – Дежерина связана с делецией D4Z4 на 4qA хромосомы 4, мутациями гена SMCHD1 в локусе 18p11.32.
Мы подготовили для Вас список исследований, которые помогут разобраться с данной проблемой:
90 рабочиx дней
Панель «Наследственные нарушения обмена веществ»
35000 руб
ПодробнееСимптоматика
Каждая форма мышечной дистрофии имеет свою особую клиническую картину, однако есть и ряд общих симптомов, характерных для данной группы заболеваний:
- постепенное снижение тонуса мышечных волокон;
- отсутствие болевого синдрома;
- сохранение чувствительности пораженных участков;
- изменение походки;
- частые падения из-за слабости мышц ног;
- изменение размеров мышц;
- постоянная усталость;
- постепенная атрофия скелетных мышц;
- потеря приобретенных физических навыков у детей.


Диагностика мышечной дистрофии
Диагностические мероприятия включают:
- оценку уровня мышечного фермента креатинкиназы крови;
- мышечную биопсию;
- электромиографию;
- генетическое тестирование.
В медико-генетическом центре «Геномед» проводятся исследования на предмет мутаций в генах, которые обуславливают мышечную дистрофию, в том числе в таких генах, как EMD, LMNA, FHL1 (миодистрофия Эмери – Дрейфуса), DMPK и ZNF9 (миотоническая форма патологии).
Мышечная дистрофия Дюшенна/Беккера. Лайонизация Х-хромосомы у девочек
Метод определения ПЦР, секвенирование Выдаётся заключение врача-генетика!
Исследуемый материал Цельная кровь (с ЭДТА)
Тип наследования.
Х-сцепленный рецессивный, т.е. им страдают почти исключительно мальчики, женщины же с поврежденным геном в одной из Х-хромосом являются носительницами МДД. Но в редких случаях миодистрофией Дюшенна могут болеть и девочки. Причинами этого могут быть преимущественная инактивация Х-хромосомы с нормальным аллелем у гетерозиготных носительниц мутантного гена дистрофина, Х-аутосомная транслокация, затрагивающая этот ген, гемизиготность по мутантному аллелю и наличие фенокопий (заболеваний, связанных с нарушением других белков, входящих в дистрофин-гликопротеиновый комплекс). Приблизительно в 2/3 случаев сын получает хромосому с повреждением от матери-носительницы, в остальных случаях заболевание возникает в результате мутации de novo в половых клетках матери или отца, либо в предшественниках этих клеток. Приблизительно 30% всех случаев заболевания связаны с возникновением свежих мутаций в гене дистрофина, а остальные 70% обусловлены носительством матерью пробанда патологической мутации в одной из Х хромосом. Считается, что 6-7% всех спорадических случаев заболевания являются следствием гонадного мозаицизма — существования в яичниках женщины нескольких генераций ооцитов с нормальными и мутантными аллелями гена дистрофина.
Приблизительно 30% всех случаев заболевания связаны с возникновением свежих мутаций в гене дистрофина, а остальные 70% обусловлены носительством матерью пробанда патологической мутации в одной из Х хромосом. Считается, что 6-7% всех спорадических случаев заболевания являются следствием гонадного мозаицизма — существования в яичниках женщины нескольких генераций ооцитов с нормальными и мутантными аллелями гена дистрофина. Гены, ответственные за развитие заболевания.
DMD (DYSTROPHIN) — ген дистрофина, находится в Х-хромосоме в регионе Хр21.2 –р21.1, состоит из 79 экзонов. У 60%-70% больных выявляются крупные делеции, захватывающие один или несколько экзонов гена и локализованные в двух «горячих» регионах — в области 5′ конца (экзоны 6-19) и 3′ конца (экзоны 40-43). У 5% больных обнаруживаются дупликации, в остальных случаях — точковые мутации. Различия в тяжести клинических проявлений при двух аллельных вариантах заболевания связывают с различиями в характере мутации в гене дистрофина. При мышечной дистрофии Дюшенна мутации в гене дистрофина приводят к сдвигу рамки считывания и преждевременной терминации трансляции, при этом синтез белка прекращается. При мышечной дистрофии Беккера структурные перестройки гена не приводят к сдвигу рамки считывания, ДНК-полимераза может «перескакивать» делетированные экзоны, что приводит к синтезу внутренне усеченного белка, который может, до некоторой степени, выполнять свои функции.
Определение заболевания.
Нейромышечное заболевание, обусловленное мутацией в гене дистрофина и приводящее к прогрессирующей дегенерации мышечных волокон.Патогенез и клиническая картина.
Основная функция дистрофина заключается в обеспечении устойчивости и эластичности мышечного волокна при последующих мышечных сокращениях. При отсутствии дистрофина вследствие мутации мембрана разрушается, в ней появляются участки некроза, что приводит к вымыванию содержимого саркоплазмы в кровяное русло. Происходит постепенная гибель мышечных волокон и замещение их соединительнотканными структурами, которые увеличивают плотность и объем мышц, вызывая феномен псевдогипертрофии. Заболевание встречается в двух клинических формах, являющихся аллельными генетическими вариантами.
Заболевание встречается в двух клинических формах, являющихся аллельными генетическими вариантами.Прогрессирующая мышечная дистрофия Дюшенна.
Заболевание проявляется в возрасте 1-5 лет, быстро прогрессирует и приводит к летальному исходу до 25 летнего возраста. Для большинства больных характерна задержка темпов раннего моторного развития. При начале самостоятельной ходьбы, в возрасте старше 14 месяцев, отмечаются частые падения, спотыкания, моторная неловкость, быстрая утомляемость. Постепенно походка становится переваливающейся, возникают затруднения при подъеме по лестнице и из положения на корточках, когда больные вынуждены использовать вспомогательные приемы Говерса («взбирание по самому себе»). На ранних стадиях заболевания обнаруживаются псевдогипертрофии мышц, возникающие за счет разрастания соединительной и жировой ткани на месте гибнущих мышечных волокон. Наиболее часто они локализуются в икроножных, дельтовидных, четырехглавых и трехглавых мышцах и создают ложное впечатление атлетического телосложения больного. По мере прогрессирования заболевания псевдогипертрофии мышц трансформируются в их гипотрофии. Распространение патологического процесса имеет восходящий характер. Первыми поражаются мышцы тазового пояса и проксимальных отделов нижних конечностей, затем мышцы плечевого пояса, спины и проксимальных отделов верхних конечностей. Уже на ранних стадиях болезни снижаются или угасают коленные рефлексы. Ахиллов рефлекс, а также сухожильные рефлексы с рук, могут длительное время оставаться сохранными. По мере развития патологического процесса в мышцах возникают вторичные деформации позвоночника (усиление лордоза и кифоза, сколиоз), грудной клетки (по типу седловидной и килевидной) и стоп, а также ретракции сухожилий с развитием контрактур в суставах. Характерным признаком является кардиомиопатия, которая проявляется симптомами гипертрофии левого желудочка и аритмией. У 25-30% больных диагностируется олигофрения в степени дебильности. Пациенты сохраняют способность к самостоятельной ходьбе до 10-12-ти летнего возраста, после чего пользуются инвалидной коляской.
Прогрессирующая мышечная дистрофия Беккера.
Наиболее часто заболевание возникает в возрастном интервале от 10 до 20 лет с появления слабости и утомляемости мышц тазового пояса и ног. Ранними симптомами у значительного числа больных бывают болезненные мышечные крампи. Клинические проявления сходны с таковыми при ПМДД, однако имеют значительно меньшую степень выраженности. Характерной особенностью ПМДБ является вовлечение в патологический процесс миокарда. Гипертрофическая или дилятационная кардиомиопатия диагностируется у 50-60% больных. В 40-50% случаев выявляются гипогенитализм и атрофия яичек. Интеллект, как правило, не страдает. Заболевание прогрессирует достаточно медленно и в большинстве случаев приводит к инвалидизации больного не ранее 40-летнего возраста.Прогрессирующая мышечная дистрофия Дюшенна у лиц женского пола.
Описаны клинические проявления ПМДД у лиц женского пола, которые являются носительницами мутации в гене дистрофина в гетерозиготном состоянии. Клинические признаки могут появиться в различные возрастные периоды, но чаще провоцируются гормональными перестройками в организме женщины (начало менструаций, беременность, климакс). Появление клинических симптомов может быть обусловлено двумя причинами: 1) наличие полной или мозаичной форм синдрома Шерешевского-Тернера; 2) феноменом несбалансированной лайонизации. На электромиограмме выявляются признаки первично-мышечного поражения в виде усиления интерференции и снижения амплитуды М-ответа. Высокую диагностическую значимость имеет определение активности фермента креатинфосфокиназы в плазме крови больного. Этот показатель у больных ПМДД в 50-100 раз превышает норму и может быть выявлен до возникновения выраженных клинических признаков. Для диагностики и дифференциальной диагностики ПМДД/ПМДБ используются иммуногистохимические методы анализа дистрофина в биоптате мышечного волокна. При использовании антисывороток на различные районы дистрофина при ПМДД иммунореактивных форм белка, как правило, не выявляется. У больных с ПМДБ наблюдается прерывистое окрашивание мышц при иммунохимическом анализе, что свидетельствует об относительной сохранности отдельных структур цитоскелета. Специфического морфологического дефекта не существует. В биоптате мышц больных выявляются изменения, характерные для группы прогрессирующих мышечных дистрофий в целом.
У больных с ПМДБ наблюдается прерывистое окрашивание мышц при иммунохимическом анализе, что свидетельствует об относительной сохранности отдельных структур цитоскелета. Специфического морфологического дефекта не существует. В биоптате мышц больных выявляются изменения, характерные для группы прогрессирующих мышечных дистрофий в целом. Частота встречаемости: Мышечная дистрофия Дюшенна (МДД): 1:2500-4000 новорожденных мальчиков. Частота МДБ (Беккера) составляет 1 на 20000 мальчиков.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Monaco, A. P.; Kunkel, L. M.: A giant locus for the Duchenne and Becker muscular dystrophy gene. Trends Genet. 3: 33-37, 1987.
- Becker, P. E.: Eine neue X-chromosomale Muskeldystrophie. Acta Psychiat. Neurol. Scand. 193: 427, 1955.
Врожденные мышечные дистрофии: классификация и диагностика | Rivier
1. Sparks S., Quijano-Roy S., Harper A. et al. Congenital muscular dystrophy overview in: Pagon RA, Bird TD, Dolan CR, et al. Gene reviews. 1993–2001.
2. Muntoni F., Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004;14(10):635–49.
3. Sparks S.E., Escolar D.M. Congenital muscular dystrophies. Handb Clin Neurol 2011;101:47–9.
4. Mercuri E., Muntoni F. The ever-expanding spectrum of congenital muscular dystrophies. Ann Neurol 2012;72(1):9–17.
5. Godfrey C., Foley A.R., Clement E. et al. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev 2011;21(3):278–85.
6. Mathews K.D., Stephan C.M., Laubenthal K. et al. Myoglobinuria and muscle pain are common in patients with limb-girdle muscular dystrophy 2I. Neurology 2011;76(2):194–5.
Mathews K.D., Stephan C.M., Laubenthal K. et al. Myoglobinuria and muscle pain are common in patients with limb-girdle muscular dystrophy 2I. Neurology 2011;76(2):194–5.
7. Clement E.M., Feng L., Mein R. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001–2008. Neuromuscul Disord 2012;22(6):522–7.
8. Hayashi Y.K., Chou F.L., Engvall E. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet 1998;19(1):94–7.
9. Hara Y., Balci-Hayta B., Yoshida-Moriguchi T. A dystroglycan mutation associated with limbgirdle muscular dystrophy. N Engl J Med 2011;364(10):939–46.
10. Godfrey C., Clement E., Mein R. et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007;130(10):2725–35.
11. Mercuri E., Messina S., Bruno C. et al. Congenital muscular dystrophies with defective glycosylation of dystroglycan: a population study. Neurology 2009;72(21):1802–9.
12. Ferreiro A., Quijano-Roy S., Pichereau C. et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet 2002;71(4):739–49.
13. Mitsuhashi S., Ohkuma A., Talim B. et al. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am J Hum Genet 2011;88(6):845–51.
Am J Hum Genet 2011;88(6):845–51.
14. Tomé F.M., Evangelista T., Leclerc A. et al. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III 1994;317(4):351–7.
15. Helbling-Leclerc A., Zhan X., Topaloglu H. et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet 1995;11(2):216–8.
16. Geranmayeh F., Clement E., Feng L.H. et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 2010;20(4):241–50.
17. Lamer S., Carlier R.Y., Pinard J.M. et al. Congenital muscular dystrophy: use of brain MR imaging findings to predict merosin deficiency. Radiology 1998;206(3):811–6.
18. Okada M., Kawahara G., Noguchi S. et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007;69(10):1035–42.
19. Allamand V., Briñas L., Richard P. et al. ColVI myopathies: where do we stand, where do we go? Skelet Muscle 2011;1:30.
20. Briñas L., Richard P., Quijano-Roy S. et al. Early onset collagen VI myopathies: Genetic and clinical correlations. Ann Neurol 2010;68(4):511–20.
21. Nadeau A., Kinali M., Main M. et al. Natural history of Ullrich congenital muscular dystrophy. Neurology 2009;73(1):25–31.
22. Mercuri E., Lampe A., Allsop J. et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. 2005;15(4):303–10.
Mercuri E., Lampe A., Allsop J. et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. 2005;15(4):303–10.
23. Quijano-Roy S., Avila-Smirnow D., Carlier R.Y. et al. Whole body muscle MRI protocol: pattern recognition in early onset NM disorders. Neuromuscul Disord 2012;22.
24. Hicks D., Lampe A.K., Barresi R. et al. A refined diagnostic algorithm for Bethlem myopathy. Neurology 2008;70(14):1192–9.
25. Moore C.J., Winder S.J. The inside and out of dystroglycan post-translational modification. Neuromuscul Disord 2012;22(11):959–65.
26. Wells L. The o-mannosylation pathway: glycosyltransferases and proteins implicated in congenital muscular dystrophy. J Biol Chem 2013;288(10):6930–5.
27. Kobayashi K., Nakahori Y., Miyake M. et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature, 1998;394(6691):388–92.
28. Yoshida A., Kobayashi K., Manya H. et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 2001;1(5):717–24.
29. Brockington M., Blake D.J., Prandini P. et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet 2001;69(6):1198–209.
30. Beltrán-Valero de Bernabé D., Currier S., Steinbrecher A. et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 2002;71(5):1033–43.
Beltrán-Valero de Bernabé D., Currier S., Steinbrecher A. et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 2002;71(5):1033–43.
31. van Reeuwijk J., Janssen M., van den Elzen C. et al. POMT2 mutations cause alphadystroglycan hypoglycosylation and Walker- Warburg syndrome. J Med Genet.2005 Dec;42(12):907–12.
32. Longman C., Brockington M., Torelli S et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet 2003;12(21):2853–61.
33. Cirak S., Foley A.R., Herrmann R. et al. ISPD gene mutations are a common cause of congenital and limb-girdle muscular dystrophies. Brain 2013;136(Pt1):269–81.
34. Barone R., Aiello C., Race V. et al. DPM2-CDG: a muscular dystrophydystroglycanopathy syndrome with severe epilepsy. Ann Neurol 2012;72(4):550–8.
35. Lefeber D.J., de Brouwer A.P., Morava E. et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PloS Genet 2011;7(12).
36. Lefeber D.J., Schönberger J., Morava E. et al. Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet 2009;85(1):76–86.
37. Willer T., Lee H., Lommel M. et al. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat Genet 2012;44(5):575–80.
Nat Genet 2012;44(5):575–80.
38. Roscioli T., Kamsteeg E.J., Buysse K. et al.Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat Genet. 2012;44(5):581–5.
39. Manzini M.C., Tambunan D.E., Hill R.S. et al. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am J Hum Genet 2012;91(3):541–7.
40. Vuillaumier-Barrot S., Bouchet-Séraphin C., Chelbi M. et al. Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly. Am J Hum Genet 2012;91(6):1135–43.
41. Stevens E., Carss K.J., Cirak S. et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of α-dystroglycan. Am J Hum Genet 2013;92(3):354–65.
42. Buysse K., Riemersma M., Powell G. et al. Missense mutations in β-1,3-Nacetylglucosaminyltransferase 1 (B3GnT1) cause Walker-Warburg syndrome. Hum Mol Genet 2013;22(9):1746–54.
43. Carss K.J., Stevens E., Foley A.R. et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of α-dystroglycan. Am J Hum Genet 2013;93(1):29–41.
44. Yang A.C., Ng B.G., Moore S.A. et al. Congenital disorder of glycosylation due to DPM1 mutations presenting with dystroglycanopathy-type congenital muscular dystrophy. Mol Genet Metab 2013; 110(3):345–51.
45. Vuillaumier-Barrot S., Quijano-Roy S., Bouchet-Seraphin C. et al. Four Caucasian patients with mutations in the fukutin gene and variable clinical phenotype. Neuromuscul Disord 2009;19(3):182–8.
Vuillaumier-Barrot S., Quijano-Roy S., Bouchet-Seraphin C. et al. Four Caucasian patients with mutations in the fukutin gene and variable clinical phenotype. Neuromuscul Disord 2009;19(3):182–8.
46. Schara U., Kress W., Bönnemann C.G. et al. The phenotype and long-term follow-up in 11 patients with juvenile selenoprotein N1-related myopathy. Eur J Paediatr Neurol 2008;12(3):224–30.
47. Scoto M., Cirak S., Mein R. et al. SEPN1-related myopathies: clinical course in a large cohort of patients. Neurology 2011;76(24):2973–8.
48. Mercuri E., Pichiecchio A., Allsop J. et al. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J Magn Reson Imaging. 2007;25(2):433–40.
49. Quijano-Roy S., Mbieleu B., Bönnemann C.G. et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol 2008;64(2):177–86.
50. Ben Yaou et al. Les Cahiers de myologie. 2010(3) :24–33.
51. Bonne G., Quijano-Roy S. Emery–Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb Clin Neurol 2013;113:1367–76.
52. Hattori A., Komaki H., Kawatani M. et al. A novel mutation in the LMNA gene causes congenital muscular dystrophy with dropped head and brain involvement. Neuromuscul Disord 2012;22(2):149–51.
53. Mercuri E., Clements E., Offiah A. et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol 2010;67(2):201–8.
Ann Neurol 2010;67(2):201–8.
54. Makri S., Clarke N.F., Richard P. et al. Germinal mosaicism for LMNA mimics autosomal recessive congenital muscular dystrophy. Neuromuscul Disord 2009;19(1):26–8.
У меня мышечная дистрофия Ландузи — Дежерина
Отношение к болезни
Иногда я думаю о том, что бы было, если бы у меня не было мышечной дистрофии. Если честно, мне до сих пор сложно поверить, что я больна. Родителям и родственникам это тоже дается непросто. Они помнят, как я бегала и прыгала, и не всегда могут осознать реальность произошедших со мной перемен.
Мне кажется, отношения с родителями у всех сложные. Моя семья не является исключением, а диагноз только все усугубляет. Дело в том, что с детства меня воспитывали так: ты либо здоровый и успешный, либо «спрыгиваешь со скалы». Поэтому мое заболевание стало большим испытанием для всех членов семьи. Особенно сложно пришлось маме, для которой мое состояние — настоящая драма. Мы еще и живем вместе, но стараемся не конфликтовать. Просто она сама по себе слабая женщина, а я сильная и волевая. Поэтому у нас иногда возникают недопонимания.
Я до сих пор вижу, как страдают некоторые родственники, когда мы встречаемся. Не представляю, что бы сказали мои одноклассники, если бы увидели меня сейчас. Правда, одна одноклассница недавно покупала у меня картину и говорила, что я и мои работы — классные. Вообще, у тех, кто никогда не видел меня здоровой, проблем с восприятием меня нет. К примеру, моя младшая двоюродная сестра вообще не парится. Хотя это вопрос приоритетов: для кого-то внешнее и физическое состояния очень важны, а для кого-то — наоборот. Особенно ярко это можно было ощутить раньше, когда человек считался самым крутым, если тусил на районе. Интернет все изменил — теперь не обязательно выходить из дома.
Если бы я знала в 13 или 16 лет, как все будет, я бы по-другому построила свою жизнь. Прежде всего я бы постаралась вдолбить своим родителям, что моя болезнь — это норма, а не наказание для семьи. Еще я бы вела себя более самостоятельно. Сейчас маме хочется меня пристроить к кому-то. Несмотря на мои заверения в собственной самодостаточности, ей кажется, что меня всю жизнь должен кто-то поддерживать.
Кроме того, я бы проще относилась к межгендерным отношениям и уже давно обзавелась бы детьми. Также я бы постаралась не перетруждаться и подготовила бы себе парашют в виде еще одной профессии. Мне довольно сложно общаться с людьми, поэтому я предпочла бы быть каким-нибудь программистом. Могла бы сидеть дома, хорошо зарабатывать, а раз в полгода ездить куда-нибудь, где хороший климат и инфраструктура. Мне хочется путешествовать, но уже несколько лет я никуда, кроме Питера, не езжу. Надеюсь скоро это исправить.
Сейчас моя главная цель — заработать денег и разъехаться с мамой. В 30 лет, наверное, буду рожать. Меня пока за такие планы в тюрьму никто не посадил и матку не вырезал. Но ребенок тоже требует подготовки и больших материальных затрат, поэтому сейчас мне надо сосредоточиться на работе.
Окулофарингеальная мышечная дистрофия
Окулофарингеальная мышечная дистрофия (ОФМД) — заболевание, которое в мире встречается в двух генетических вариантах: аутосомно-рецессивном и аутосомно-доминантном. Эти варианты болезни являются аллельными и обусловлены различными мутациями в одном гене. Клинические варианты описаны в зависимости от типа наследования. Несмотря на то что, согласно литературным данным, ОФМД относится к более поздним формам мышечных дистрофий (4 – 5 –е десятилетие), имеется клинический пример раннего дебюта ОФМД у пациента в возрасте 23 года, с установленным диагнозом в 31 год.
| Симптомы ОФМД: гипомимия, двусторонний птоз век, наружная офтальмоплегия, «локальная миопатия», поражающая глазодвигательные и поднимающие веки мышцы, а также мышцы-констрикторы глотки (заболевание опасно развитием нарастающей дисфагии), дисфония При длительном течении заболевания — заметная гипотрофия нескольких групп мышц: лица, плечевого пояса, в т. Средний возраст манифестации: 49 ± 1,42 лет |
 ч. спины, конечностей. Часто появляются бронхо-легочные осложнения.
ч. спины, конечностей. Часто появляются бронхо-легочные осложнения.Особенность окулофарингеальной миодистрофии — наличие в ядрах нитевидных трубочек диаметром 8,5 нм. Аутосомно-доминантный и аутосомно-рецессивный варианты болезни обусловлены мутациями в одном и том же гене — РАВР2 (полиаденилсвязывающем протеине-2; OMIM 602 279), локализованном в области 14q11.2-q13. Основной тип мутаций — короткая экспансия тринуклеотидного повтора GCG в кодирующей части гена. В норме число повторов не превышает 6, однако у 2 % здоровых людей число повторов может достигать 7, что расценивается как проявление нормального полиморфизма. У больных с окулофарингеальной миопатией число повторов увеличено до 8–13.
Электрофореграмма сиквенса GCG – повторов в 1 экзоне гена PABPN1 Результаты электрофореза ДНК на ДНК-анализаторе ABIPRISM 310Тяжесть проявления заболевания зависит от количества повторов. Антиципация, обусловленная увеличением количества повторов, не характерна. Возникновение аутосомно-рецессивного варианта обусловлено гомозиготностью по GCG7-повтору, который является примером аллели-модификатора. Наиболее тяжелый фенотип наблюдался у компаунд-гетерозигот GCG9/GCG7, а также гомозигот по GCG9-повторам. Патогенетически белок РАВР2 является высоконсервативным и содержится в ядре, где участвует в полиаденилировании мРНК. GCG-повторы кодируют включение полиаланинового тракта вблизи N-конца мутантного белка. Считается, что образующиеся в ядре нитевидные структуры представлены удлиненными нитями мутантного белка. Заболевание особенно часто встречается у франко-язычных канадцев и в латиноамериканских семьях на юго-западе США. Оно описано также в большой еврейской семье восточноевропейского происхождения. На сегодняшний день для профилактики ОФМД возможна дородовая диагностика с использованием методов ДНК-анализа.
ДНК-диагностика ОФМД в 8% ПААГ: (М — маркер PUC19/MspI) 1, 4 — генотип 6/6 GCG-повторов (здоровые). 2, 3 — генотип 6/10 GCG-повторов (больные). |

Опасность окулофарингеальной миодистрофии состоит в прогрессировании с нарастающей дисфагией и требует применения методов паллиативной неврологии. Паллиативная неврология предусматривает зондовое питание или наложение стомы. Эффективного лечения на данный момент нет. Описаны методики рассечения перстнеглоточной мышцы для улучшения глотания, но не предотвращения аспирации. Если птоз мешает зрению, используют специальные скотчевые наклейки на веки, проволочные держатели век, которые крепятся к оправе очков, либо, если нет выраженной слабости мимических мышц, прибегают к хирургическому лечению.
Аутосомно-доминантный вариант заболевания (впервыеописан Victor и соавтор. в 1962 году у 9 членов одной семьи из трех поколений ). Первые симптомы возникают на 4–5-м десятилетиях жизни и в большинстве случаев характеризуются сочетанием дисфагии с прогрессирующим птозом верхних век. По мере прогрессирования заболевания отмечается распространение симптомов мышечной слабости на мышцы плечевого и тазового поясов. Также писана (Satoyoshi & Kinoshita, 1977) семья с аутосомно-доминантной сегрегацией окулофарингеальной миопатии, характеризующейся значительной генерализацией процесса по мере течения болезни. У наблюдаемых больных мышечная слабость распространялась на мышцы лица, шеи, дистальных отделов конечностей, а также анального сфинктера. Описаны единичные больные с наличием пигментной дегенерации сетчатки. Большинство авторов эту форму аутосомно-доминантной миопатии относят к довольно редким, возникающим в зрелом возрасте и медленнотекущим заболеваниям, считают, что клинически болезнь проявляет себя как локальная миопатия. Поражаются мышцы, осуществляющие движения глазных яблок, и мышца, поднимающая верхнее веко. Фарингеальные расстройства обусловлены включением в процесс констрикторов глотки, что затрудняет глотание. Типична симметричность процесса. Начальные признаки болезни появляются в возрасте 30–40 лет: двустороннее опущение верхнего века при ограничении движения глазных яблок. Как правило, на диплопию больные не жалуются. Объяснение этому находят в медленном и симметричном развитии парезов глазодвигательных мышц. Значительно утяжеляют заболевание и ухудшают прогноз фарингеальные симптомы. Начинаясь с дисфагии, они имеют тенденцию к нарушению функций (афагии). Следует иметь в виду существование окулярной миопатии, при которой фарингеальные расстройства не выражены. Этот вариант миопатии рассматривается одними исследователями как самостоятельное заболевание, другими- как дебют окулофарингеальной миопатии.
Начальные признаки болезни появляются в возрасте 30–40 лет: двустороннее опущение верхнего века при ограничении движения глазных яблок. Как правило, на диплопию больные не жалуются. Объяснение этому находят в медленном и симметричном развитии парезов глазодвигательных мышц. Значительно утяжеляют заболевание и ухудшают прогноз фарингеальные симптомы. Начинаясь с дисфагии, они имеют тенденцию к нарушению функций (афагии). Следует иметь в виду существование окулярной миопатии, при которой фарингеальные расстройства не выражены. Этот вариант миопатии рассматривается одними исследователями как самостоятельное заболевание, другими- как дебют окулофарингеальной миопатии.
Аутосомно-рецессивный вариант заболевания (впервые описан Fried и соавт. в 1975 году у двух сестер, родившихся от кровнородственного брака). Для этой формы болезни характерно более раннее начало и вовлечение в процесс дистальных групп мышц конечностей. При (ЭМГ) электромиографическом обследовании больных с ОФМД определяют первично-мышечный характер поражения. При морфологическом исследовании выявляют нитевидные образования в ядрах скелетных мышц. Эти нити имеют ветвящуюся трубчатую структуру и иногда поперечно исчерчены. Наряду с этим отмечаются атрофические изменения в мышечных волокнах 1-го типа. При электронном микроскопировании обнаруживается увеличение размеров митохондрий с наличием в них крестовидных включений. Также могут быть обнаружены вакуоли, при электронной микроскопии в них видны обрывки мембран, скопления гликогена и другие неспецифичные остатки лизосомного происхождения.
Распространенность ОФМД у якутов в 10 раз выше распространенности описанной в мире. ОФМД получила накопление на территории улусов центральной Якутии и вилюйских улусов, особенно в Усть-Алданском улусе и городе Якутске.Выявлены мажорные мутации, характерные для якутской популяции: экспансия (GCG)10 в гене PÁBPN1 для окулофарингеальной миодистрофии. Распространенность аутосомно-доминантной окулофарингеальной миодистрофии в PC (Я) среди якутов составляет 11,1 на 100 тыс. населения с преимущественным накоплением в центральной и вилюйской группах улусов Якутии. Единственная молекулярно-генетическая причина всех случаев ОФМД в якутской популяции — мутация (GCG)m в 1 экзоне гена PABPN1. Выявленный единственный гаплотип является гаплотипом основателя.
населения с преимущественным накоплением в центральной и вилюйской группах улусов Якутии. Единственная молекулярно-генетическая причина всех случаев ОФМД в якутской популяции — мутация (GCG)m в 1 экзоне гена PABPN1. Выявленный единственный гаплотип является гаплотипом основателя.
Литература по ОФМД в якутской популяции:
- Внедрение ДНК-диагностики окулофарингеальной миодистрофии в практику медико-генетического консультирования Республики Саха (Якутия) // Куртанов Х.А., Максимова Н.Р., Стапанова С.К. и др. // Якутский медицинский журнал. 2008. №4. С. 43-46.
- Генетико-эпидемиологические и социально-экономические аспекты наследственной этноспецифической патологии в Якутии // Максимова Н.Р., Сухомясова A.JI., Гуринова Е.Е. и др. // Медицинская генетика. 2008. Т. 7. №10. С. 35-43.
- ДНК-диагностика моногенных заболеваний в Республике Саха (Якутия)//Максимова Н.Р., Сухомясова A.JL, Томский М.И. // Илин. 2009. №3. С. 54-56.
- Клинико-генеалогическая и молекулярно-генетическая характеристика окулофарингеальной миодистрофии в Республике Саха (Якутия)//Максимова Н.Р., Николаева И.А., Коротов М.Н. и др. // Журнал неврологии и психиатрии им. С.С. Корсакова. 2008. Т. 108. №6. С. 52-60.
- Клинико-генеалогическая и молекулярно-генетическая характеристика этноспецифических форм наследственной патологии у якутов // Максимова Надежда Романовна //автореф. дисс. на соиск. уч. степ. доктора медицинских наук // Томск – 2009
- Клинико-генеалогическое и молекулярно-генетическое исследование окулофарингеальной миодистрофии в Республике Саха (Якутия) // Максимова Н.Р., Николаева И.А., Коротов М.Н. и др. / C6.VIII научно-практической конференции «Генетика человека и патология». Томск: Изд-во «Печатная мануфактура», 2007. С. 160-161.
- Молекулярно-генетические методы диагностики наследственных моногенных болезней в РБ№1-НЦМ PC (Я) //Степанова С.К., Кононова С.К., Федорова С.А., Максимова Н.Р. и др. // Тез. межрегиональной научно-практической конференции «Молекулярно-клеточные аспекты патологии человека на Севере». Якутск: ЯНЦ СО РАМН, 2007. С. 49-51.
- Наследственные болезни нервной системы в Республике Саха (Якутия) // Николаева И.А., Коротов М.Н., Гуринова Е.Е., Степанова С.К., Максимова Н.Р. и др. // Якутский медицинский журнал. 2009. №2. С. 52-54.
- Наследственные болезни у якутов // В.П.Пузырев, Н.Р.Максимова
- Окулофарингеальная миодистрофия в Республике Саха (Я) // Максимова Н.Р., Коротов М.Н., Николаева И.А. и др. // Тез. III международной научно-практической конференции «Проблемы вилюйского энцефаломиелита и других нейродегенеративных заболеваний в Якутии». Якутск: «Копиртех-сервис», 2006. С. 22-23.
- миодистрофия в Республике Саха (Якутия): клинические и молекулярно-генетические аспекты // Максимова Н.Р., Николаева И.А., Коротов М.Н. и др. // Тез. П межрегиональной научно-практической конференции «Экология и здоровье человека на Севере». Якутск: ЯНЦ СО РАМН, 2007. С. 34.
- Полиморфизм локуса ОФМД в популяциях Якутии // Куртанов Х.А., Максимова Н.Р., Марусин A.B., Степанов В.А. // Якутский медицинский журнал. 2009. №2. С. 54-58.
- Этноспецифические наследственные болезни у якутов // Максимова Н.Р., Пузырев В.П / Тез. межрегиональной научно-практической конференции «Здоровье детей на Севере». Якутск: ЯНЦ СО РАМН, 2008. С. 91-94.
- Этноспецифическая наследственная патология в РС (Я)// Максимова Н.Р., Сухомясова А.Л., Ноговицына А.Н., Пузырев В.П.
- Литература по ОФМД в других популяциях: ДНК-диагностика случая окулофарингеальной миодистрофии в России // Чухрова А.Л., Федотов В.П., Поляков А.В. // Медицинская генетика, 2003 г., Т.2. №10 «Тезисы Всероссийской научно-практической конф. «Соврем. достиж. клинич. генетики», Москва, 25-27 ноября 2003 г., стр. 446
- Клинико-молекулярно-генетическая классификация мышечных дистрофий (научный обзор с комментариями // Казаков В.М. // Неврол. журнал. — 2001. — № 3. — С. 47-52.
- Молекулярная неврология. Часть 1. Заболевания нервно-мышечной системы. // Горбунова В.Н., Савельева-Васильева Е.А., Красильников В.В. // СПб.: Интермедика, 2000. — 320 с.
- Ранняя клинико-инструментальная диагностика и терапия быстро- и медленнопрогрессирующих мышечных дистрофий и амиотрофий // 3. Евтушенко С.К., Шаймурзин М.Р., Евтушенко О.С., Евтушенко Л.Ф., Дегонская Е.В., Евтушенко И.С., Сохань Д.А.. // Международный неврологический журнал. — 2007. — № 4(14). — С. 47.
- Справочник по формулированию клинического диагноза болезней нервной системы / Под ред. В.Н. Штока, О.С. Левина. — М.: ООО «Медицинское информационное агентство», 2006. — 520 с.
- Brais B., Bouchard J.-P., Xie Y.-G., Rochefort D.L., Chretien N., Tome F.M.S., Lafreniere R.G., Rommens J.M., Uyama E., Nohira O., Blumen S., Korcyn A.D., Heutink P., Mathieu J., Duran-ceau A., Codere F., Fardeau M., Rouleau G.A. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy // Nature Genet. — 1998. — 18. — 164-167.
- Brais B., Xie Y.-G., Sanson M., Morgan K., Weissenbach J., Korczyn A.D., Blumen S.C., Fardeau M., Tome F.M.S., Bou-chard J.-P., Rouleau G.A. The oculopharyngeal muscular dystrophy locus maps to the region of the cardiac alpha and beta myosin heavy chain genes on chromosome 14q11.2-q13 // Hum. Molec. Genet. — 1995. — 4. — 429-434.
- Fried K., Arlozorov A., Spria R. Autosomal recessive oculopharyngeal muscular dystrophy // J. Med. Genet. — 1975. — 12. — 416-418.
- Goh K.J., Wong K.T., Nishino I., Minami N., Nonaka I. Oculopharyngeal muscular dystrophy with PABPN1 mutation in a Chinese Malaysian woman // Neuromusc. Disord. 2005. — 15. — 262-264.
- Hino H., Araki K., Uyama E., Takeya M., Araki M., Yoshinobu K., Miike K., Kawazoe Y., Maeda Y., Uchino M., Yamamura K. Myopathy phenotype in transgenic mice expressing mutated PABPN1 as a model of oculopharyngeal muscular dystrophy // Hum. Molec. Genet. — 2004. — 13. — 181-190.
- Neuromuscular disorders: gene location // Neuromusc. Disord. — 1999. — Vol. 9, № 5. — P. 1-8.
- Robinson D.O., Wills A.J., Hammans S.R., Read S.P., Sillibourne J. Oculopharyngeal muscular dystrophy: a point mutation which mimics the effect of the PABPN1 gene triplet repeat expansion mutation // J. Med. Genet. — 2006. — 43. — e23 (Note: Electronic Article).
- Victor M., Hayes R., Adams R.D. Oculopharyngeal muscular dystrophy. A familial disease of late life characterized by dysphagia and progressive ptosis of the eyelids // New Eng. J. Med. — 1962. — 267. — 1267-1272.
 Якутск: ЯНЦ СО РАМН, 2007. С. 49-51.
Якутск: ЯНЦ СО РАМН, 2007. С. 49-51. // Горбунова В.Н., Савельева-Васильева Е.А., Красильников В.В. // СПб.: Интермедика, 2000. — 320 с.
// Горбунова В.Н., Савельева-Васильева Е.А., Красильников В.В. // СПб.: Интермедика, 2000. — 320 с. — 1999. — Vol. 9, № 5. — P. 1-8.
— 1999. — Vol. 9, № 5. — P. 1-8.| Тип | Возраст начала болезни | Симптомы, скорость прогрессирования и ожидаемая продолжительность жизни |
|---|---|---|
| Беккер | от подросткового до раннего взросления | Возраст в дебюте Возраст в дебюте Симптомы почти идентичны Дюшенну, но менее серьезны; прогрессирует медленнее, чем по Дюшенну; дожить до среднего возраста.Как и в случае с Дюшенном, болезнь почти всегда ограничивается мужчинами. |
| Врожденный | рождение | Возраст в дебюте Возраст в дебюте Симптомы включают общую мышечную слабость и возможные деформации суставов; болезнь медленно прогрессирует; сокращенная продолжительность жизни. |
| Дюшенн | От 2 до 6 лет | Возраст в дебюте Возраст в дебюте Симптомы включают общую мышечную слабость и истощение; поражает таз, верхнюю часть рук и верхнюю часть ног; в конечном итоге задействует все произвольные мышцы; выживание после 20 лет — редкость. Встречается только у мальчиков. Очень редко может повлиять на женщину, у которой гораздо более легкие симптомы и лучший прогноз. Встречается только у мальчиков. Очень редко может повлиять на женщину, у которой гораздо более легкие симптомы и лучший прогноз. |
| Дистальный | От 40 до 60 лет | Возраст в дебюте Возраст в дебюте Симптомы включают слабость и истощение мышц рук, предплечий и голеней; прогрессирование медленное; редко приводит к полной нетрудоспособности. |
| Эмери-Драйфус | от детства до раннего подросткового возраста | Возраст в дебюте Возраст в дебюте Симптомы включают слабость и истощение мышц плеча, предплечья и голени; суставные деформации распространены; прогрессирование медленное; внезапная смерть может наступить из-за проблем с сердцем. |
| Лицево-лопаточно-плечевой | от детства к ранним взрослым | Возраст в дебюте Возраст в дебюте Симптомы включают слабость и слабость лицевых мышц с некоторым истощением плеч и предплечий; прогрессирование медленное с периодами быстрого ухудшения; продолжительность жизни может составлять многие десятилетия после начала болезни. |
| Ремень для конечностей | от позднего детства до среднего возраста | Возраст в дебюте Возраст в дебюте Симптомы включают слабость и истощение, поражающие в первую очередь плечевой и тазовый пояс; прогрессирование медленное; смерть обычно наступает из-за сердечно-легочных осложнений. |
| Миотонический | От 20 до 40 лет | Возраст в дебюте Возраст в дебюте Симптомы включают слабость всех групп мышц, сопровождающуюся замедленным расслаблением мышц после сокращения; поражает в первую очередь лицо, ступни, руки и шею; прогрессирование идет медленно, иногда от 50 до 60 лет. |
| Окулофарингеальный | От 40 до 70 лет | Возраст в дебюте Возраст в дебюте Симптомы поражают мышцы век и горла, вызывая ослабление мышц горла, что со временем приводит к неспособности глотать и истощению из-за недостатка пищи; прогрессирование идет медленно. |
Мышечная дистрофия: симптомы, диагностика и лечение
Что такое мышечная дистрофия?
Мышечная дистрофия — это группа заболеваний, при которых со временем мышцы становятся слабее и менее гибкими. Это вызвано проблемой в генах, которые контролируют, как тело поддерживает здоровье мышц. У некоторых людей болезнь начинается в раннем детстве. У других нет никаких симптомов, пока они не станут подростками или взрослыми людьми среднего возраста.
Каким образом мышечная дистрофия влияет на вас или вашего ребенка, зависит от ее вида.Состояние большинства людей со временем ухудшается, и некоторые люди могут потерять способность ходить, говорить или заботиться о себе. Но это случается не со всеми. Другие люди могут жить долгие годы с легкими симптомами.
Существует более 30 видов мышечной дистрофии, и каждый из них отличается в зависимости от:
- Гены, вызывающие ее
- Мышцы, на которые она влияет
- Возраст, когда симптомы впервые появляются
- Как быстро болезнь ухудшается
Люди обычно болеют одной из девяти основных форм болезни:
- Мышечная дистрофия Дюшенна (МДД) является наиболее распространенной формой. В основном она поражает мальчиков и начинается в возрасте от 3 до 5 лет.
- Мышечная дистрофия Беккера похожа на мышечную дистрофию Дюшена, за исключением более легкой степени. Он также поражает мальчиков, но симптомы проявляются позже — в возрасте от 11 до 25 лет.
- Миотоническая мышечная дистрофия — наиболее распространенная форма у взрослых. Люди, у которых он есть, не могут расслабить мышцы после сокращения. Это может повлиять как на мужчин, так и на женщин, и обычно начинается, когда людям за двадцать.
- Врожденная мышечная дистрофия начинается при рождении или вскоре после этого.
- Мышечная дистрофия конечностей и пояса часто начинается в подростковом или 20-летнем возрасте.
- Facioscapulohumeral мышечная дистрофия поражает мышцы лица, плеч и предплечий. Это может повлиять на кого угодно, от подростков до взрослых в возрасте от 40 лет.
- Дистальная мышечная дистрофия поражает мышцы рук, ног, кистей и стоп. Обычно она возникает в более позднем возрасте, в возрасте от 40 до 60 лет.
- Окулофарингеальная мышечная дистрофия начинается у человека в возрасте от 40 до 50 лет.Это вызывает слабость мышц лица, шеи и плеч и опущение век (птоз) с последующим затруднением глотания (дисфагия).
- Мышечная дистрофия Эмери-Дрейфуса поражает главным образом мальчиков, обычно начиная с 10 лет. Люди с этой формой часто имеют проблемы с сердцем наряду с мышечной слабостью.
 В основном она поражает мальчиков и начинается в возрасте от 3 до 5 лет.
В основном она поражает мальчиков и начинается в возрасте от 3 до 5 лет.Существует множество методов лечения, которые могут помочь сохранить мышцы сильными и гибкими, и ученые тоже ищут новые. Важно получить необходимое лечение и найти поддержку.
Причины
Мышечная дистрофия может передаваться в семье, или вы можете быть первым в своей семье, у кого она есть. Это состояние вызвано проблемами в ваших генах.
Гены содержат информацию, необходимую вашим клеткам для выработки белков, контролирующих все функции организма. Когда у гена есть проблема, ваши клетки могут вырабатывать неправильный белок, неправильное его количество или поврежденный белок.
Когда у гена есть проблема, ваши клетки могут вырабатывать неправильный белок, неправильное его количество или поврежденный белок.
Вы можете получить мышечную дистрофию, даже если ни один из ваших родителей не болел.Это происходит, когда один из ваших генов дефектен сам по себе. Но так бывает редко.
У людей с мышечной дистрофией поврежденные гены — это те, которые вырабатывают белки, которые поддерживают здоровье и силу мышц. Например, люди с мышечной дистрофией Дюшенна или Беккера вырабатывают слишком мало белка, называемого дистрофином, который укрепляет мышцы и защищает их от травм.
Симптомы
Для большинства типов мышечной дистрофии симптомы начинают проявляться в детстве или в подростковом возрасте.Как правило, дети с этим заболеванием:
- Часто падают
- Имеют слабые мышцы
- Имеют мышечные судороги
- Проблемы с подъемом, подъемом по лестнице, бегом или прыжками
- Ходите на цыпочках или переваливайте
Некоторые также будут иметь следующие симптомы:
- Искривленный позвоночник (так называемый сколиоз)
- Обвисшие веки
- Проблемы с сердцем
- Проблемы с дыханием или глотанием
- Проблемы со зрением
- Слабость мышц лица
Диагностика
Вашему врачу необходимо будет проверить различные части тела вашего ребенка, чтобы узнать, есть ли у них мышечная дистрофия.Они начнут с общего медицинского осмотра. Они также зададут вам вопросы об истории болезни вашей семьи и о симптомах, которые вы замечаете у своего ребенка. Они могут спросить:
- Какие мышцы доставляют им проблемы?
- Им трудно ходить или заниматься своими обычными делами?
- Как давно это происходит?
- Есть ли у кого-нибудь в вашей семье мышечная дистрофия? Какой?
Они также могут задать вам вопросы о том, как ваш ребенок играет, двигается и говорит, а также как он ведет себя дома и в школе.
Продолжение
Врач может использовать различные тесты для проверки условий, которые могут вызвать мышечную слабость.
- Анализы крови. Они проверяют уровень определенных ферментов, выделяемых мышцами при их повреждении.
- Электромиография, или ЭМГ . Ваш врач наложит маленькие иглы, называемые электродами, на разные части тела вашего ребенка и попросит его медленно согнуть и расслабить мышцы. Электроды прикреплены проводами к устройству, измеряющему электрическую активность.
- Биопсия мышцы. С помощью иглы врач удаляет небольшой кусочек мышечной ткани вашего ребенка. Они будут смотреть на это под микроскопом, чтобы определить, какие белки отсутствуют или повреждены. Этот тест может показать, какой тип мышечной дистрофии может быть у вашего ребенка.
- Тесты мышечной силы, рефлексов и координации. Это помогает врачам исключить другие проблемы с их нервной системой.
- Электрокардиограмма или ЭКГ . Он измеряет электрические сигналы, исходящие от сердца, и сообщает, насколько быстро бьется сердце вашего ребенка и его ритм здоровый.
- Визуализация может показать качество и количество мышц в теле вашего ребенка. Они могут получить:
- МРТ или магнитно-резонансная томография. Он использует мощные магниты и радиоволны, чтобы делать снимки их органов.
- Ультразвук, , который использует звуковые волны для создания изображений внутренней части тела.
Врачи также могут проанализировать образец своей крови, чтобы найти гены, вызывающие мышечную дистрофию. Генетические тесты могут помочь диагностировать заболевание, но они также важны для людей с семейным анамнезом заболевания, которые планируют создать семью.Вы можете поговорить со своим врачом или консультантом по генетике, чтобы узнать, что результаты этого теста значат для вас и ваших детей.
Вопросы для врача
Вам нужно узнать как можно больше о состоянии вашего ребенка, чтобы узнать, как он может оставаться максимально здоровым. Вы можете спросить:
- Какая у них мышечная дистрофия?
- Нужны ли им еще тесты?
- Нужны ли нам другие врачи?
- Как болезнь повлияет на их жизнь?
- Какие виды лечения доступны?
- Как они будут себя чувствовать?
- Что я могу сделать, чтобы их мышцы оставались сильными?
- Существуют ли какие-либо клинические испытания, которые были бы им полезны?
- Заболеют ли другие мои дети мышечной дистрофией?
Лечение
Сейчас лекарства от болезни нет. Но существует множество методов лечения, которые могут улучшить симптомы и облегчить жизнь вам и вашему ребенку.
Но существует множество методов лечения, которые могут улучшить симптомы и облегчить жизнь вам и вашему ребенку.
Ваш врач порекомендует лечение в зависимости от типа мышечной дистрофии вашего ребенка. Вот некоторые из них:
- Физическая терапия использует различные упражнения и растяжки, чтобы мышцы оставались сильными и гибкими.
- Трудотерапия учит вашего ребенка, как максимально использовать то, на что способны его мышцы. Терапевты также могут показать им, как пользоваться инвалидными колясками, скобами и другими приспособлениями, которые могут помочь им в повседневной жизни.
- Логопед научит их более простым способам говорить, если у них слабые мышцы горла или лица.
- Респираторная терапия может помочь, если у вашего ребенка проблемы с дыханием. Они узнают, как облегчить дыхание, или попросят в помощь машины.
- Лекарства могут облегчить симптомы. Они включают:
- Этеплирсен (Exondys 51), голодирсен (Vyondys53) и витоларсен (Вилтепсо) для лечения МДД. Это инъекционные препараты, которые помогают лечить людей с определенной мутацией гена, приводящей к МДД, в частности, за счет увеличения выработки дистрофина.Поговорите с врачом вашего ребенка о возможных побочных эффектах.
- Противосудорожные препараты , уменьшающие мышечные спазмы.
- Лекарства от кровяного давления , которые помогают при проблемах с сердцем.
- Лекарства, подавляющие иммунную систему организма , , называемые иммунодепрессантами; они могут замедлить повреждение мышечных клеток.
- Стероиды , такие как преднизон и дефказакорт (Эмфлаза), которые замедляют повреждение мышц и могут помочь вашему ребенку лучше дышать.Они могут вызывать серьезные побочные эффекты, такие как слабость костей и повышенный риск инфекций.
- Креатин, химическое вещество, обычно содержащееся в организме, которое может помочь снабжать энергией мышцы и повышать силу у некоторых людей. Спросите врача, подходят ли ему эти добавки.
- Хирургия может помочь с различными осложнениями мышечной дистрофии, такими как проблемы с сердцем или проблемы с глотанием.
 Спросите врача, подходят ли ему эти добавки.
Спросите врача, подходят ли ему эти добавки.Ученые также ищут новые способы лечения мышечной дистрофии в клинических испытаниях.Эти испытания проверяют новые лекарства, чтобы убедиться, что они безопасны и работают. Часто они позволяют людям попробовать новое лекарство, доступное далеко не каждому. Ваш врач может сказать вам, подходит ли одно из этих испытаний для вашего ребенка.
Забота о ребенке
Трудно, когда ваш ребенок теряет силы и не может делать то, что могут делать другие дети. Мышечная дистрофия — это проблема, но она не должна мешать вашему ребенку получать удовольствие от жизни.
Есть много вещей, которые вы можете сделать, чтобы помочь им почувствовать себя сильнее и получить от жизни максимум удовольствия.
- Ешьте правильно. Здоровая, хорошо сбалансированная диета полезна для вашего ребенка в целом. Это также важно для поддержания здорового веса, который может облегчить проблемы с дыханием и другие симптомы. Если им сложно жевать или глотать, посоветуйтесь с диетологом о продуктах, которые, возможно, будет легче съесть.
- Оставайся активным. Физические упражнения могут улучшить мышечную силу вашего ребенка и улучшить его самочувствие. Попробуйте занятия с низкой нагрузкой, например плавание.
- Высыпайтесь. Спросите своего врача или терапевта о некоторых кроватях или подушках, которые могут сделать вашему ребенку более комфортное и отдохнувшее.
- Используйте подходящие инструменты. Инвалидные коляски, костыли или электросамокаты могут помочь вашему ребенку, если у него проблемы с ходьбой.
Болезнь, скорее всего, сильно повлияет на вашу семью. Помните, что это нормально — обращаться за помощью к врачу, психологу, семье или друзьям с любым стрессом, грустью или гневом, которые вы можете испытывать. Группы поддержки также являются хорошим местом для общения с другими людьми, которые жили с мышечной дистрофией.Они могут помочь вашему ребенку общаться с такими же людьми, как они, и дать вам и вашей семье совет и понимание.
Группы поддержки также являются хорошим местом для общения с другими людьми, которые жили с мышечной дистрофией.Они могут помочь вашему ребенку общаться с такими же людьми, как они, и дать вам и вашей семье совет и понимание.
Чего ожидать
Мышечная дистрофия у всех разная. Некоторые дети могут очень медленно терять мышечную силу, что дает им и их семьям время, чтобы приспособиться к изменениям. Другим будет хуже быстрее. Многим людям с этим заболеванием в какой-то момент понадобятся инвалидные коляски и помощь в повседневной жизни, но это не всегда так.
Поговорите со своим врачом о мышечной дистрофии вашего ребенка.Вместе вы сможете составить для них наилучший план лечения и получить необходимую поддержку для своей семьи.
Получение поддержки
Чтобы узнать больше о мышечной дистрофии или найти группу поддержки в вашем районе, посетите веб-сайт Ассоциации мышечной дистрофии.
Информационная страница о мышечной дистрофии | Национальный институт неврологических расстройств и инсульта
Определение
Лечение
Прогноз
Клинические испытания
Организации
Публикации
Определение
Мышечные дистрофии (МД) — это группа из более чем 30 генетических заболеваний, характеризующихся прогрессирующей слабостью и дегенерацией скелетных мышц, контролирующих движения.Некоторые формы MD наблюдаются в младенчестве или детстве, в то время как другие могут появиться только в среднем возрасте или позже. Расстройства различаются по распределению и степени мышечной слабости (некоторые формы MD также влияют на сердечную мышцу), возрасту начала, скорости прогрессирования и типу наследования.
Расстройства различаются по распределению и степени мышечной слабости (некоторые формы MD также влияют на сердечную мышцу), возрасту начала, скорости прогрессирования и типу наследования.
Duchenne MD является наиболее распространенной формой MD и в первую очередь поражает мальчиков. Это вызвано отсутствием дистрофина, белка, участвующего в поддержании целостности мышц. Начало заболевания составляет от 3 до 5 лет, и заболевание быстро прогрессирует.Большинство мальчиков не могут ходить к 12 годам, и позже им понадобится респиратор, чтобы дышать. Девочки в этих семьях имеют 50-процентный шанс унаследовать и передать дефектный ген своим детям. Мальчики с Беккера MD (очень похожи на MD Duchenne, но менее тяжелы) имеют дефектный или недостаточный дистрофин.
Facioscapulohumeral MD обычно начинается в подростковом возрасте. Это вызывает прогрессирующую слабость в мышцах лица, рук, ног, а также в области плеч и груди. Он прогрессирует медленно и может варьировать по симптомам от легкой до инвалидизирующей.
Миотонический MD является наиболее распространенной формой заболевания у взрослых и характеризуется продолжительными мышечными спазмами, катарактой, сердечными и эндокринными нарушениями. У людей с миотонической БМ длинные тонкие лица, опущенные веки и лебединая шея.
×
Определение
Мышечные дистрофии (МД) — это группа из более чем 30 генетических заболеваний, характеризующихся прогрессирующей слабостью и дегенерацией скелетных мышц, контролирующих движения.Некоторые формы MD наблюдаются в младенчестве или детстве, в то время как другие могут появиться только в среднем возрасте или позже. Расстройства различаются по распределению и степени мышечной слабости (некоторые формы MD также влияют на сердечную мышцу), возрасту начала, скорости прогрессирования и типу наследования.
Duchenne MD является наиболее распространенной формой MD и в первую очередь поражает мальчиков. Это вызвано отсутствием дистрофина, белка, участвующего в поддержании целостности мышц. Начало заболевания составляет от 3 до 5 лет, и заболевание быстро прогрессирует.Большинство мальчиков не могут ходить к 12 годам, и позже им понадобится респиратор, чтобы дышать. Девочки в этих семьях имеют 50-процентный шанс унаследовать и передать дефектный ген своим детям. Мальчики с Беккера MD (очень похожи на MD Duchenne, но менее тяжелы) имеют дефектный или недостаточный дистрофин.
Facioscapulohumeral MD обычно начинается в подростковом возрасте. Это вызывает прогрессирующую слабость в мышцах лица, рук, ног, а также в области плеч и груди. Он прогрессирует медленно и может варьировать по симптомам от легкой до инвалидизирующей.
Миотонический MD является наиболее распространенной формой заболевания у взрослых и характеризуется продолжительными мышечными спазмами, катарактой, сердечными и эндокринными нарушениями. У людей с миотонической БМ длинные тонкие лица, опущенные веки и лебединая шея.
Лечение
Не существует специального лечения, чтобы остановить или обратить вспять любую форму БМ.Лечение может включать физиотерапию, респираторную терапию, логопедию, ортопедические приспособления, используемые для поддержки, и корректирующую ортопедическую хирургию. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило инъекции препаратов голодирсена и вилтоларсена для лечения пациентов с мышечной дистрофией Дюшенна (МДД), у которых есть подтвержденная мутация гена дистрофина, поддающаяся пропуску экзона 53. Эти препараты способствуют выработке дистрофина. По оценкам, около 8% пациентов с МДД имеют эту мутацию. Введение препарата казимерсен одобрено для пациентов с подтвержденной мутацией гена DMD, которая поддается пропуску экзона 45. FDA также одобрило три заявки на первые генерики финголимода (Gilenya) для лечения рецидивирующих форм РС у взрослых. Лекарственная терапия других форм БМ включает кортикостероиды для замедления мышечной дегенерации, противосудорожные препараты для контроля судорог и некоторой мышечной активности, иммунодепрессанты для отсрочки повреждения умирающих мышечных клеток и антибиотики для борьбы с респираторными инфекциями.Некоторым людям может быть полезна трудотерапия и вспомогательные технологии. Некоторым пациентам может потребоваться вспомогательная вентиляция легких для лечения слабости дыхательных мышц и кардиостимулятор при сердечных нарушениях.
Введение препарата казимерсен одобрено для пациентов с подтвержденной мутацией гена DMD, которая поддается пропуску экзона 45. FDA также одобрило три заявки на первые генерики финголимода (Gilenya) для лечения рецидивирующих форм РС у взрослых. Лекарственная терапия других форм БМ включает кортикостероиды для замедления мышечной дегенерации, противосудорожные препараты для контроля судорог и некоторой мышечной активности, иммунодепрессанты для отсрочки повреждения умирающих мышечных клеток и антибиотики для борьбы с респираторными инфекциями.Некоторым людям может быть полезна трудотерапия и вспомогательные технологии. Некоторым пациентам может потребоваться вспомогательная вентиляция легких для лечения слабости дыхательных мышц и кардиостимулятор при сердечных нарушениях.
×
Лечение
Не существует специального лечения, чтобы остановить или обратить вспять любую форму БМ.Лечение может включать физиотерапию, респираторную терапию, логопедию, ортопедические приспособления, используемые для поддержки, и корректирующую ортопедическую хирургию. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило инъекции препаратов голодирсена и вилтоларсена для лечения пациентов с мышечной дистрофией Дюшенна (МДД), у которых есть подтвержденная мутация гена дистрофина, поддающаяся пропуску экзона 53. Эти препараты способствуют выработке дистрофина. По оценкам, около 8% пациентов с МДД имеют эту мутацию.Введение препарата казимерсен одобрено для пациентов с подтвержденной мутацией гена DMD, которая поддается пропуску экзона 45. FDA также одобрило три заявки на первые генерики финголимода (Gilenya) для лечения рецидивирующих форм РС у взрослых. Лекарственная терапия других форм БМ включает кортикостероиды для замедления мышечной дегенерации, противосудорожные препараты для контроля судорог и некоторой мышечной активности, иммунодепрессанты для отсрочки повреждения умирающих мышечных клеток и антибиотики для борьбы с респираторными инфекциями. Некоторым людям может быть полезна трудотерапия и вспомогательные технологии. Некоторым пациентам может потребоваться вспомогательная вентиляция легких для лечения слабости дыхательных мышц и кардиостимулятор при сердечных нарушениях.
Некоторым людям может быть полезна трудотерапия и вспомогательные технологии. Некоторым пациентам может потребоваться вспомогательная вентиляция легких для лечения слабости дыхательных мышц и кардиостимулятор при сердечных нарушениях.
Определение
Мышечные дистрофии (МД) — это группа из более чем 30 генетических заболеваний, характеризующихся прогрессирующей слабостью и дегенерацией скелетных мышц, контролирующих движения.Некоторые формы MD наблюдаются в младенчестве или детстве, в то время как другие могут появиться только в среднем возрасте или позже. Расстройства различаются по распределению и степени мышечной слабости (некоторые формы MD также влияют на сердечную мышцу), возрасту начала, скорости прогрессирования и типу наследования.
Duchenne MD является наиболее распространенной формой MD и в первую очередь поражает мальчиков. Это вызвано отсутствием дистрофина, белка, участвующего в поддержании целостности мышц. Начало заболевания составляет от 3 до 5 лет, и заболевание быстро прогрессирует.Большинство мальчиков не могут ходить к 12 годам, и позже им понадобится респиратор, чтобы дышать. Девочки в этих семьях имеют 50-процентный шанс унаследовать и передать дефектный ген своим детям. Мальчики с Беккера MD (очень похожи на MD Duchenne, но менее тяжелы) имеют дефектный или недостаточный дистрофин.
Facioscapulohumeral MD обычно начинается в подростковом возрасте. Это вызывает прогрессирующую слабость в мышцах лица, рук, ног, а также в области плеч и груди. Он прогрессирует медленно и может варьировать по симптомам от легкой до инвалидизирующей.
Миотонический MD является наиболее распространенной формой заболевания у взрослых и характеризуется продолжительными мышечными спазмами, катарактой, сердечными и эндокринными нарушениями. У людей с миотонической БМ длинные тонкие лица, опущенные веки и лебединая шея.
Лечение
Не существует специального лечения, чтобы остановить или обратить вспять любую форму БМ. Лечение может включать физиотерапию, респираторную терапию, логопедию, ортопедические приспособления, используемые для поддержки, и корректирующую ортопедическую хирургию.Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило инъекции препаратов голодирсена и вилтоларсена для лечения пациентов с мышечной дистрофией Дюшенна (МДД), у которых есть подтвержденная мутация гена дистрофина, поддающаяся пропуску экзона 53. Эти препараты способствуют выработке дистрофина. По оценкам, около 8% пациентов с МДД имеют эту мутацию. Введение препарата казимерсен одобрено для пациентов с подтвержденной мутацией гена DMD, которая поддается пропуску экзона 45.FDA также одобрило три заявки на первые генерики финголимода (Gilenya) для лечения рецидивирующих форм РС у взрослых. Лекарственная терапия других форм БМ включает кортикостероиды для замедления мышечной дегенерации, противосудорожные препараты для контроля судорог и некоторой мышечной активности, иммунодепрессанты для отсрочки повреждения умирающих мышечных клеток и антибиотики для борьбы с респираторными инфекциями. Некоторым людям может быть полезна трудотерапия и вспомогательные технологии. Некоторым пациентам может потребоваться вспомогательная вентиляция легких для лечения слабости дыхательных мышц и кардиостимулятор при сердечных нарушениях.
Прогноз
Прогноз для людей с MD варьируется в зависимости от типа и прогрессирования заболевания. Некоторые случаи могут быть легкими и прогрессировать очень медленно в течение нормальной продолжительности жизни, в то время как другие вызывают сильную мышечную слабость, функциональную инвалидность и потерю способности ходить. Некоторые дети с MD умирают в младенчестве, в то время как другие доживают до взрослого возраста, имея лишь среднюю степень инвалидности.
Некоторые дети с MD умирают в младенчестве, в то время как другие доживают до взрослого возраста, имея лишь среднюю степень инвалидности.
х
Прогноз
Прогноз для людей с MD варьируется в зависимости от типа и прогрессирования заболевания. Некоторые случаи могут быть легкими и прогрессировать очень медленно в течение нормальной продолжительности жизни, в то время как другие вызывают сильную мышечную слабость, функциональную инвалидность и потерю способности ходить.Некоторые дети с MD умирают в младенчестве, в то время как другие доживают до взрослого возраста, имея лишь среднюю степень инвалидности.
Прогноз
Прогноз для людей с MD варьируется в зависимости от типа и прогрессирования заболевания. Некоторые случаи могут быть легкими и прогрессировать очень медленно в течение нормальной продолжительности жизни, в то время как другие вызывают сильную мышечную слабость, функциональную инвалидность и потерю способности ходить.Некоторые дети с MD умирают в младенчестве, в то время как другие доживают до взрослого возраста, имея лишь среднюю степень инвалидности.
Определение
Мышечные дистрофии (МД) — это группа из более чем 30 генетических заболеваний, характеризующихся прогрессирующей слабостью и дегенерацией скелетных мышц, контролирующих движения. Некоторые формы MD наблюдаются в младенчестве или детстве, в то время как другие могут появиться только в среднем возрасте или позже.Расстройства различаются по распределению и степени мышечной слабости (некоторые формы MD также влияют на сердечную мышцу), возрасту начала, скорости прогрессирования и типу наследования.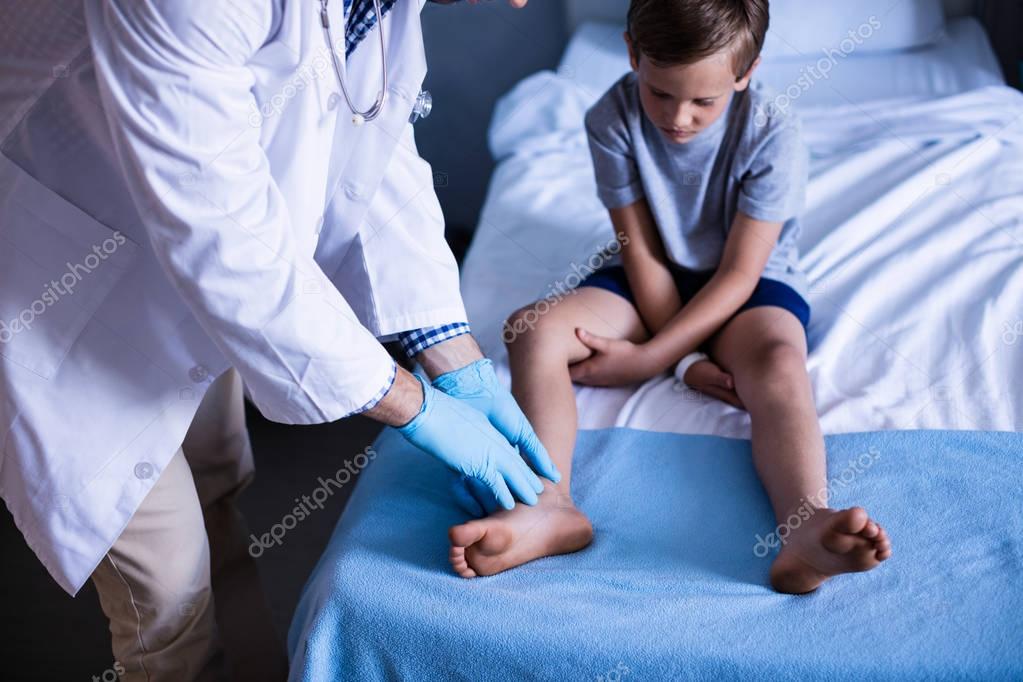
Duchenne MD является наиболее распространенной формой MD и в первую очередь поражает мальчиков. Это вызвано отсутствием дистрофина, белка, участвующего в поддержании целостности мышц. Начало заболевания составляет от 3 до 5 лет, и заболевание быстро прогрессирует. Большинство мальчиков не могут ходить к 12 годам, и позже им понадобится респиратор, чтобы дышать.Девочки в этих семьях имеют 50-процентный шанс унаследовать и передать дефектный ген своим детям. Мальчики с Беккера MD (очень похожи на MD Duchenne, но менее тяжелы) имеют дефектный или недостаточный дистрофин.
Facioscapulohumeral MD обычно начинается в подростковом возрасте. Это вызывает прогрессирующую слабость в мышцах лица, рук, ног, а также в области плеч и груди. Он прогрессирует медленно и может варьировать по симптомам от легкой до инвалидизирующей.
Миотонический MD является наиболее распространенной формой заболевания у взрослых и характеризуется продолжительными мышечными спазмами, катарактой, сердечными и эндокринными нарушениями.У людей с миотонической БМ длинные тонкие лица, опущенные веки и лебединая шея.
Лечение
Не существует специального лечения, чтобы остановить или обратить вспять любую форму БМ. Лечение может включать физиотерапию, респираторную терапию, логопедию, ортопедические приспособления, используемые для поддержки, и корректирующую ортопедическую хирургию. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило инъекции препаратов голодирсена и вилтоларсена для лечения пациентов с мышечной дистрофией Дюшенна (МДД), у которых есть подтвержденная мутация гена дистрофина, поддающаяся пропуску экзона 53.Эти препараты способствуют выработке дистрофина. По оценкам, около 8% пациентов с МДД имеют эту мутацию. Введение препарата казимерсен одобрено для пациентов с подтвержденной мутацией гена DMD, которая поддается пропуску экзона 45. FDA также одобрило три заявки на первые генерики финголимода (Gilenya) для лечения рецидивирующих форм РС у взрослых. Лекарственная терапия других форм БМ включает кортикостероиды для замедления мышечной дегенерации, противосудорожные препараты для контроля судорог и некоторой мышечной активности, иммунодепрессанты для отсрочки повреждения умирающих мышечных клеток и антибиотики для борьбы с респираторными инфекциями.Некоторым людям может быть полезна трудотерапия и вспомогательные технологии. Некоторым пациентам может потребоваться вспомогательная вентиляция легких для лечения слабости дыхательных мышц и кардиостимулятор при сердечных нарушениях.
FDA также одобрило три заявки на первые генерики финголимода (Gilenya) для лечения рецидивирующих форм РС у взрослых. Лекарственная терапия других форм БМ включает кортикостероиды для замедления мышечной дегенерации, противосудорожные препараты для контроля судорог и некоторой мышечной активности, иммунодепрессанты для отсрочки повреждения умирающих мышечных клеток и антибиотики для борьбы с респираторными инфекциями.Некоторым людям может быть полезна трудотерапия и вспомогательные технологии. Некоторым пациентам может потребоваться вспомогательная вентиляция легких для лечения слабости дыхательных мышц и кардиостимулятор при сердечных нарушениях.
Прогноз
Прогноз для людей с MD варьируется в зависимости от типа и прогрессирования заболевания. Некоторые случаи могут быть легкими и прогрессировать очень медленно в течение нормальной продолжительности жизни, в то время как другие вызывают сильную мышечную слабость, функциональную инвалидность и потерю способности ходить.Некоторые дети с MD умирают в младенчестве, в то время как другие доживают до взрослого возраста, имея лишь среднюю степень инвалидности.
Какие исследования проводятся?
NINDS поддерживает широкую программу исследований по MD. Цели этих исследований — понять MD и разработать методы диагностики, лечения, профилактики и, в конечном итоге, излечения расстройства.
NINDS является членом Координационного комитета по мышечной дистрофии (MDCC).Для получения дополнительной информации посетите: https://mdcc.nih.gov/
Информация из MedlinePlus Национальной медицинской библиотеки
Мышечная дистрофия
Организации пациентов
Центры по контролю и профилактике заболеваний (CDC)
Министерство здравоохранения и социальных служб США
1600 Клифтон-роуд
Атланта
GA
Атланта, Джорджия 30333
Тел . : 800-311-3435; 404-639-3311; 404-639-3543
: 800-311-3435; 404-639-3311; 404-639-3543
Коалиция по лечению Кальпаина 3 (C3)
15 Compo Parkway
Вестпорт
CT
Westport, CT 06880
Тел .: 203-829-9656
Лечение ВМД (врожденная мышечная дистрофия)
217 E.Карсон-стрит, # 1014
Лейквуд
CA
Lakewood, CA
Тел .: 562-444-5656
CureDuchenne
— 1400 Перепелиная улица (индекс
)Люкс 110
Ньюпорт-Бич
CA
Ньюпорт-Бич, Калифорния 92660
Тел .: 949-872-2552
Общество лицево-лопаточно-плечевой мышечной дистрофии (ФСГ)
450 Бедфорд-стрит,
Лексингтон
MA
Lexington, MA 02420
Тел .: 718-301-6060
Фонд джайнов
9706 Четвертая авеню, NE
Люкс 101
Сиэтл
WA
Сиэтл, Вашингтон 98115
Тел .: 425-882-1440
Ассоциация мышечной дистрофии
Национальный офис — 222 S.Риверсайд Плаза
161 Н. Кларк, офис 3550
Чикаго
IL
Чикаго, Иллинойс 60601
Тел .: 800-572-1717
Тональный крем для миотонической дистрофии
663 Тринадцатая улица,
Люкс 101
Окленд
CA
Окленд, Калифорния 94612
Тел .: 415-800-7777; 866-968-6422
Национальный институт артрита и костно-мышечной и кожных заболеваний (NIAMS)
Национальные институты здравоохранения, DHHS
31 Центр Др., Rm. 4C02 МСК 2350
Bethesda
MD
Bethesda, Мэриленд 20892-2350
Тел .: 301-496-8190; 877-22-НИАМС (226-4267)
Национальный институт детского здоровья и развития человека (NICHD)
Национальные институты здравоохранения, DHHS
31 Center Drive, Rm. 2А32 МСК 2425
2А32 МСК 2425
Bethesda
MD
Bethesda, Мэриленд 20892-2425
Тел .: 301-496-5133
Родительский проект мышечной дистрофии (PPMD)
401 Hackensack Avenue, 9-й этаж
Хакенсак
NJ
Hackensack, NJ 07601
Тел .: 800-714-5437
Публикации
Информационный листMuscular Dystrophy (MD), составленный Национальным институтом неврологических расстройств и инсульта (NINDS).
Организации пациентов
Центры по контролю и профилактике заболеваний (CDC)
Министерство здравоохранения и социальных служб США
1600 Клифтон-роуд
Атланта
GA
Атланта, Джорджия 30333
Тел .: 800-311-3435; 404-639-3311; 404-639-3543
Коалиция по лечению Кальпаина 3 (C3)
15 Compo Parkway
Вестпорт
CT
Westport, CT 06880
Тел .: 203-829-9656
Лечение ВМД (врожденная мышечная дистрофия)
217 E.Карсон-стрит, # 1014
Лейквуд
CA
Lakewood, CA
Тел .: 562-444-5656
CureDuchenne
— 1400 Перепелиная улица (индекс
)Люкс 110
Ньюпорт-Бич
CA
Ньюпорт-Бич, Калифорния 92660
Тел .: 949-872-2552
Общество лицево-лопаточно-плечевой мышечной дистрофии (ФСГ)
450 Бедфорд-стрит,
Лексингтон
MA
Lexington, MA 02420
Тел . : 718-301-6060
: 718-301-6060
Фонд джайнов
9706 Четвертая авеню, NE
Люкс 101
Сиэтл
WA
Сиэтл, Вашингтон 98115
Тел .: 425-882-1440
Ассоциация мышечной дистрофии
Национальный офис — 222 S.Риверсайд Плаза
161 Н. Кларк, офис 3550
Чикаго
IL
Чикаго, Иллинойс 60601
Тел .: 800-572-1717
Тональный крем для миотонической дистрофии
663 Тринадцатая улица,
Люкс 101
Окленд
CA
Окленд, Калифорния 94612
Тел .: 415-800-7777; 866-968-6422
Национальный институт артрита и костно-мышечной и кожных заболеваний (NIAMS)
Национальные институты здравоохранения, DHHS
31 Центр Др., Rm. 4C02 МСК 2350
Bethesda
MD
Bethesda, Мэриленд 20892-2350
Тел .: 301-496-8190; 877-22-НИАМС (226-4267)
Национальный институт детского здоровья и развития человека (NICHD)
Национальные институты здравоохранения, DHHS
31 Center Drive, Rm. 2А32 МСК 2425
Bethesda
MD
Bethesda, Мэриленд 20892-2425
Тел .: 301-496-5133
Родительский проект мышечной дистрофии (PPMD)
401 Hackensack Avenue, 9-й этаж
Хакенсак
NJ
Hackensack, NJ 07601
Тел .: 800-714-5437
Мышечная дистрофия: симптомы, причины, лечение
Обзор
Что такое мышечная дистрофия?
Мышечная дистрофия относится к группе из более чем 30 наследственных (генетических) заболеваний, вызывающих мышечную слабость. Эти состояния являются разновидностью миопатии, заболевания скелетных мышц. Со временем мышцы сжимаются и ослабевают, что влияет на вашу способность ходить и выполнять повседневные действия, например чистить зубы. Заболевание также может поразить ваше сердце и легкие.
Эти состояния являются разновидностью миопатии, заболевания скелетных мышц. Со временем мышцы сжимаются и ослабевают, что влияет на вашу способность ходить и выполнять повседневные действия, например чистить зубы. Заболевание также может поразить ваше сердце и легкие.
Некоторые формы мышечной дистрофии проявляются при рождении или развиваются в детстве. Некоторые формы развиваются позже, в зрелом возрасте. В настоящее время лекарства нет.
Насколько распространена мышечная дистрофия?
Мышечная дистрофия — редкое заболевание.
Кто может получить мышечную дистрофию?
Мышечная дистрофия часто передается по наследству. Ребенок, у которого есть родитель с мышечной дистрофией, может унаследовать мутировавший (измененный) ген, вызывающий мышечную дистрофию. У некоторых людей есть мутировавший ген, но нет мышечной дистрофии. Эти здоровые взрослые (носители) могут передать мутировавший ген своему ребенку, у которого может развиться болезнь.
Какие бывают типы мышечной дистрофии?
Существует более 30 различных типов мышечной дистрофии.Некоторые из наиболее распространенных форм включают:
- Мышечная дистрофия Дюшенна (МДД): Это заболевание, как правило, поражает мальчиков в возрасте от 2 до 5 лет, но могут заболеть и девочки. Вы можете заметить, что вашему малышу трудно бегать, ходить или прыгать. По мере прогрессирования заболевания оно может поражать сердце и легкие ребенка. МДД — наиболее распространенная форма мышечной дистрофии. От него страдают примерно шесть из 100 000 детей в Северной Америке и Европе.
- Мышечная дистрофия Беккера (МПК): МПК является второй по распространенности мышечной дистрофией.Симптомы МПК могут появиться в любое время в возрасте от 5 до 60 лет, но обычно появляются в подростковом возрасте. Мужчины чаще заболевают МПК. Заболевание поражает мышцы бедра, бедра и плеча, а в конечном итоге и сердце. Приблизительно у одного из 18 000–30 000 мальчиков в США развивается МПК.
- Facioscapulohumeral мышечная дистрофия (FSHD): FSHD является третьей по распространенности мышечной дистрофией. Заболевание поражает мышцы лица, лопаток и предплечий. Симптомы обычно появляются в возрасте до 20 лет.Около четырех человек из 100 000 в США имеют эту форму.
- Врожденные мышечные дистрофии (ВМД): Врожденные состояния, такие как ВМД, присутствуют при рождении. У младенца могут быть слабые мышцы, искривленный позвоночник и слишком жесткие или расшатанные суставы. У детей с ВМД могут быть проблемы с обучением, судороги и проблемы со зрением.
- Мышечная дистрофия Эмери-Драйффа (EDMD): Это заболевание, как правило, поражает детей. К 10 годам появляются такие симптомы, как слабость плеч, предплечий и икроножных мышц.EDMD также влияет на сердце.
- Конечностно-поясная мышечная дистрофия (LGMD): Это заболевание поражает мышцы, расположенные ближе всего к телу, включая плечи и бедра. От него страдают люди любого возраста. Примерно двое из 100000 человек в США имеют LGMD.
- Миотоническая дистрофия: У людей с миотонией возникают проблемы с расслаблением мышц. Например, вам может быть трудно отпустить руку любимого человека. Заболевание поражает также сердце и легкие.Это заболевание, как правило, встречается у взрослых европейского происхождения и встречается примерно у 10 из 100 000 человек.
- Окулофарингеальная мышечная дистрофия (OPMD): Эта редкая форма мышечной дистрофии ослабляет мышцы век и горла. Такие симптомы, как опущение век (птоз) и затрудненное глотание (дисфагия), часто появляются между 40 и 60 годами. Примерно один из 100 000 человек имеет OPMD.
Симптомы и причины
Что вызывает мышечную дистрофию?
Генетические мутации или изменения вызывают большинство форм мышечной дистрофии.Один или оба родителя могут передать дефектный ген своему ребенку, даже если у родителя нет этого заболевания. В редких случаях мышечная дистрофия развивается спонтанно, то есть причина неизвестна.
Каковы симптомы мышечной дистрофии?
Мышечная слабость — главный симптом мышечной дистрофии. В зависимости от вида заболевание поражает разные мышцы и части тела. Другие признаки мышечной дистрофии включают:
- Увеличенные икроножные мышцы.
- Затруднения при ходьбе или беге.
- Необычная походка (например, при ходьбе).
- Проблемы с глотанием.
- Проблемы с сердцем, такие как аритмия и сердечная недостаточность (кардиомиопатия).
- Нарушения обучаемости.
- Жесткие или свободные суставы.
- Мышечная боль.
- Искривление позвоночника (сколиоз).
- Проблемы с дыханием.
Диагностика и тесты
Как диагностируется мышечная дистрофия?
Если ваш лечащий врач подозревает мышечную дистрофию, вы или ваш ребенок можете пройти один или несколько из следующих диагностических тестов:
- Анализ крови на фермент и белок проверяет повышенный уровень фермента, называемого креатинкиназой.Высокий уровень может указывать на повреждение мышц, вызванное мышечной дистрофией.
- Электромиография (ЭМГ) измеряет электрическую активность мышц и нервов.
- Биопсия мышцы позволяет выявить клеточные изменения в мышечной ткани.
- Генетические тесты выявляют генные мутации, связанные с мышечной дистрофией.
Ведение и лечение
Как лечить или лечить мышечную дистрофию?
Исследователи все еще ищут способ вылечить мышечную дистрофию.Симптомы заболевания со временем ухудшаются, но эти методы лечения могут помочь:
- Лечебные процедуры и лечебные процедуры укрепляют и растягивают мышцы. Эти методы лечения могут помочь вам сохранить функцию и диапазон движений.
- Логопед помогает тем, у кого проблемы с глотанием.
- Кортикостероиды, такие как преднизон и дефлазакорт, могут замедлять прогрессирование заболевания.
- Хирургия снимает напряжение сокращенных мышц и исправляет искривление позвоночника (сколиоз).
- Сердечные устройства, например, кардиостимуляторы, используются для лечения нарушений сердечного ритма и сердечной недостаточности.
- Медицинские устройства, такие как ходунки и инвалидные коляски, могут улучшить подвижность и предотвратить падения.
- Средства для ухода за респираторными заболеваниями, например приспособления от кашля и респираторы, способствующие дыханию.
Каковы осложнения мышечной дистрофии?
Мышечная дистрофия поражает мышцы, сердце и легкие. По мере прогрессирования болезни вы можете быть более подвержены:
- Проблемы с сердцем, такие как аритмия и сердечная недостаточность.
- Респираторные инфекции, включая пневмонию.
- Проблемы с дыханием.
- Удушье.
Как мышечная дистрофия влияет на беременность?
Женщины с мышечной дистрофией могут иметь здоровую беременность. Поскольку мышечная дистрофия передается по наследству, вы можете поговорить с генетическим консультантом до зачатия. Во время беременности ваш лечащий врач будет внимательно следить за вашим здоровьем, чтобы помочь вам избежать следующих осложнений:
- Повышенная мышечная слабость и ограниченная подвижность из-за увеличения веса.
- Проблемы с дыханием из-за дополнительного давления на легкие.
- Напряжение сердца.
- Выкидыш (прерывание беременности до полного развития ребенка).
- Преждевременные роды до 37 недели беременности.
- Ребенок с низким весом при рождении, весом менее 5 фунтов 8 унций.
Профилактика
Как предотвратить мышечную дистрофию?
К сожалению, вы ничего не можете сделать, чтобы предотвратить мышечную дистрофию.Если у вас есть заболевание, эти шаги помогут вам улучшить качество жизни:
- Соблюдайте здоровую диету, чтобы предотвратить недоедание.
- Пейте много воды, чтобы избежать обезвоживания и запора.
- Делайте как можно больше упражнений.
- Поддерживайте здоровый вес, чтобы предотвратить ожирение.
- Бросьте курить, чтобы защитить свои легкие и сердце.
- Сделайте прививки от гриппа и пневмонии.
Перспективы / Прогноз
Каков прогноз (перспективы) для людей с мышечной дистрофией?
Мышечная дистрофия — заболевание прогрессирующее.Симптомы со временем ухудшаются. Физическая и трудотерапия, а также медицинские устройства, такие как ходунки, могут помочь вам сохранить подвижность и независимость как можно дольше. Медицинские специалисты проводят лечение и могут дать рекомендации по защите сердца и легких.
Жить с
Когда мне позвонить врачу?
Вам следует позвонить своему врачу, если у вас мышечная дистрофия и вы испытываете:
- Признаки респираторной инфекции.
- Затрудненное глотание или удушье.
- Учащенное сердцебиение или боль в груди.
- Мышечная боль.
Какие вопросы я должен задать своему врачу?
Если у вас мышечная дистрофия, вы можете спросить своего врача:
- Есть ли у моих детей риск мышечной дистрофии?
- Какие шаги я могу предпринять, чтобы предотвратить передачу гена мышечной дистрофии моим детям?
- Следует ли мне пройти генетическое тестирование?
- Как мышечная дистрофия повлияет на качество моей жизни?
- Укорачивает ли моя жизнь мышечная дистрофия?
- Какие шаги я могу предпринять, чтобы замедлить прогрессирование болезни?
- Какие изменения я могу ожидать по мере прогрессирования болезни?
- Если у близкого человека мышечная дистрофия, следует ли мне пройти генетическое тестирование, чтобы узнать, несу ли я мутацию гена?
- Стоит ли обращать внимание на признаки осложнений?
Записка из клиники Кливленда
Трудно жить с таким заболеванием, как мышечная дистрофия, которое медленно ограничивает вашу способность двигаться и функционировать.Утрата независимости может быть эмоционально тяжелой для вас и ваших близких. Не бойтесь сообщить своему врачу, если вас не устраивает ваша ситуация. Антидепрессанты, консультации и группы поддержки предлагают помощь и надежду. Ваш лечащий врач также может порекомендовать методы лечения и медицинские устройства, которые помогут вам как можно дольше сохранять независимость.
Мышечная дистрофия Дюшенна — NORD (Национальная организация по редким заболеваниям)
УЧЕБНИКИ
Behrman RE, Kliegman RM, Jenson HB, eds.Учебник педиатрии Нельсона. 17-е изд. Филадельфия, Пенсильвания: Эльзевьер Сондерс; 2005: 2060-9.
Acsadi G. Мышечная дистрофия Дюшенна. Руководство NORD по редким заболеваниям. Филадельфия, Пенсильвания: Липпинкотт Уильямс и Уилкинс: 2003: 623.
СТАТЬИ ИЗ ЖУРНАЛА
Foster K, Foster H, Dickson JG. Прогресс и перспективы генной терапии: мышечная дистрофия Дюшенна. Gene Ther. 2006; 13: 1677-85.
Flanigan KM, von Niederhausern A, Dunn DM, et al., Быстрый прямой анализ последовательности гена дистрофина.Am J Hum Genet. 2003; 72: 931-9.
Дэвис Дж. Э., Винокур Т. С., Аарон М. Ф. и др. Кардиомиопатия у носителя мышечной дистрофии Дюшенна. J Пересадка сердца и легких. 2001; 20: 781-4.
Fenichel GM, Griggs RC, Kissel J, et al., Рандомизированное исследование эффективности и безопасности оксандролона при лечении мышечной дистрофии Дюшенна. Неврология. 2001; 56: 1075-9.
Дубовиц В. Мышечные дистрофии — ясность или хаос? N Engl J Med. 1997; 336: 650-651.
Боланд Б.Дж., Зильберт П.Л., Грувер Р.В., Воллан П.К., Сильверштейн М.Д.Скелетная, сердечная недостаточность и недостаточность гладких мышц при мышечной дистрофии Дюшенна. Pediat Neurol. 1996; 14: 7-12.
Бушби К.М.Д., Эпплтон Р., Андерсон Л.В.Б. и др. Делеционный статус и интеллектуальные нарушения при мышечной дистрофии Дюшенна. Dev Med Child Neurol. 1995; 37: 260-269.
Barbujani G, Russo A, Danieli GA, et al. Анализ сегрегации 1885 семей с МДД: значительное отклонение от ожидаемой доли спорадических случаев. Hum Genet. 1990; 84: 522-526.
Mendell JR, Moxley RT, Griggs RC, et al.Рандомизированное двойное слепое шестимесячное исследование преднизона при мышечной дистрофии Дюшенна. N Engl J Med. 1989; 320: 1592-1597.
ИНТЕРНЕТ
Darras BT, Miller DT, Urion DK. Дистрофинопатии. 5 сентября 2000 г. [Обновлено 26 ноября 2014 г.]. В: Pagon RA, Adam MP, Ardinger HH, et al., Редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2016 гг. Доступно по адресу: http://www.ncbi.nlm.nih.gov/books/NBK1119/ По состоянию на 21 июня 2016 г.
Онлайн-менделевское наследование в человеке (OMIM).Университет Джона Хопкинса. Мышечная дистрофия по типу Дюшенна; DMD. Запись №: 310200. Последнее обновление 6.06.2016. Доступно по адресу: http://omim.org/entry/310200 По состоянию на 21 июня 2016 г.
Мышечная дистрофия | Сидарс-Синай
Не то, что вы ищете?Обзор
Мышечная дистрофия — это группа генетических заболеваний, вызывающих прогрессирующую слабость мышц тела. Некоторые типы мышечной дистрофии проявляются симптомами в раннем детстве, а другие — в зрелом возрасте.Различные группы мышц также могут быть затронуты в зависимости от типа мышечной дистрофии.
Мышечная дистрофия Дюшенна — наиболее распространенная форма мышечной дистрофии у детей. Обычно он появляется у мальчиков 3–6 лет и быстро ухудшается.
Мышечная дистрофия Беккера имеет симптомы, аналогичные мышечной дистрофии Дюшенна. Однако симптомы чаще всего начинаются в подростковом возрасте до 25 лет и медленно прогрессируют.
Симптомы
Различные типы мышечной дистрофии начинают проявлять симптомы в разном возрасте, и, в зависимости от типа мышечной дистрофии, поражаются разные группы мышц по всему телу.
Симптомы могут включать:
- Прогрессирующая мышечная слабость и истощение (атрофия)
- Ходьба вразвалкой
- Сложный подъем по лестнице
- Затруднение при вставании из положения лежа или сидя
- Повторное падение
- Искривление позвоночника
- Истощение мышц бедра
- Аномальное увеличение икр
- Проблемы с дыханием или глотанием
- Конечности втягиваются внутрь и фиксируются в положении
- Увеличение сердца
Помимо мышечных дистрофий Дюшенна и Беккера, существуют и другие, менее распространенные типы:
- Фациоскапуло-плечевая мышечная дистрофия (синдром Ландузи-Дежерина), поражающая в основном мышцы лица, плеч и рук
- Мышечная дистрофия пояснично-конечностного отдела с первым поражением бедер и плеч
- Миотоническая мышечная дистрофия (MMD или болезнь Штейнерта), которая вызывает неспособность расслабить мышцы и обычно сначала поражает лицевые мышцы
- Окулофарингеальная мышечная дистрофия, обычно возникающая в среднем возрасте со слабостью мышц глаз, лица и горла, вызывающая опущение век и проблемы с глотанием
Причины и факторы риска
Дефектные гены являются причиной мышечной дистрофии.Специфическое генное заболевание было обнаружено для наиболее распространенных мышечных дистрофий. Например, дефектный ген дистрофина, белка, который помогает сохранить неповрежденными мышечные клетки, является причиной мышечной дистрофии Дюшенна.
Что касается менее распространенных форм мышечной дистрофии, исследователи все еще пытаются найти конкретный дефект гена, который вызывает заболевание. Хотя большинство форм мышечной дистрофии передаются по наследству, некоторые из них возникают в результате спонтанной генной мутации без какой-либо семейной истории болезни.
Диагностика
Чтобы диагностировать любую форму мышечной дистрофии, врач тщательно изучит историю болезни и проведет медицинский осмотр. Также можно заказать некоторые диагностические тесты.
Анализ крови покажет, вытекает ли фермент креатинкиназа из мышечных клеток, вызывая аномально высокий уровень в крови. Хотя высокий уровень креатинкиназы в крови не обязательно подтверждает наличие у пациента мышечной дистрофии, это признак мышечной болезни.
Электромиография и исследования нервной проводимости позволяют измерять электрическую активность мышц и нервов для выявления наличия, локализации и степени заболеваний, которые могут повредить мышечную ткань.
Биопсия мышцы, при которой небольшой кусочек мышцы удаляется для исследования под микроскопом, обычно проводится для подтверждения состояния. Под микроскопом мышца после положительной биопсии обычно показывает мертвую ткань и аномально большие мышечные волокна. На поздних стадиях мышечной дистрофии жир и другие ткани заменяют мертвую мышечную ткань.
Генетическое тестирование может быть выполнено для определения генных мутаций, вызвавших мышечную дистрофию.
Лечение
В настоящее время не существует известного лекарства от мышечной дистрофии. Лечебная физкультура и упражнения помогают предотвратить постоянное сокращение мышц вокруг суставов и избежать искривления позвоночника. Иногда требуется операция, чтобы освободить напряженные болезненные мышцы. Дыхательные упражнения помогают отсрочить ослабление дыхательных мышц.
Некоторые симптомы можно вылечить, а прогрессирование можно замедлить с помощью лекарств. Преднизон, мощный кортикостероидный препарат, в настоящее время используется для временного снятия мышечной слабости и замедления их повреждения, а также для улучшения дыхательной функции. При некоторых типах мышечной дистрофии могут возникнуть проблемы с сердцем, которые можно лечить с помощью лекарств или кардиостимулятора.
Исследователи изучают генную терапию, которая позволила бы мышцам вырабатывать дистрофин (для мышечных дистрофий Дюшенна и Беккера), а также другие методы лечения, чтобы найти лекарство от всех мышечных дистрофий.
Не то, что вы ищете?Мышечная дистрофия — NHS
Мышечные дистрофии (МД) — это группа наследственных генетических состояний, которые постепенно вызывают ослабление мышц, что приводит к возрастающему уровню инвалидности.
MD — это прогрессирующее состояние, что означает, что со временем оно ухудшается. Он часто начинается с воздействия на определенную группу мышц, а затем затрагивает более широкие мышцы.
Некоторые типы MD в конечном итоге влияют на сердце или мышцы, используемые для дыхания, после чего состояние становится опасным для жизни.
От MD неизлечимо, но лечение может помочь справиться со многими симптомами.
Что вызывает мышечную дистрофию?
MD вызван изменениями (мутациями) генов, отвечающих за структуру и функционирование мышц человека.
Мутации вызывают изменения в мышечных волокнах, которые мешают мышцам функционировать.Со временем это приводит к увеличению инвалидности.
Мутации часто передаются по наследству от родителей. Если в вашем семейном анамнезе есть MD, ваш терапевт может направить вас на генетическое тестирование и консультацию, чтобы оценить ваш риск развития этого заболевания или рождения ребенка с MD, а также обсудить доступные вам варианты.
Узнайте больше о причинах MD и генетическом тестировании на MD.
Виды мышечной дистрофии
Существует много разных типов БМ, каждый из которых имеет несколько разные симптомы.Не все типы вызывают тяжелую инвалидность, и многие не влияют на продолжительность жизни.
Некоторые из наиболее распространенных типов MD включают:
- Дюшенн MD — одна из наиболее частых и тяжелых форм, обычно поражает мальчиков в раннем детстве; люди с этим заболеванием обычно доживают до 20-30 лет
- миотоническая дистрофия — разновидность МД, которая может развиться в любом возрасте; На продолжительность жизни не всегда влияет, но люди с тяжелой формой миотонической дистрофии могли сократить жизнь
- facioscapulohumeral MD — тип MD, который может развиться в детстве или в зрелом возрасте; прогрессирует медленно и обычно не опасно для жизни
- Becker MD — близкий родственник Duchenne MD, но развивается позже в детстве и протекает в меньшей степени; продолжительность жизни обычно не так сильно влияет на продолжительность жизни
- limb-girdle MD — группа состояний, которые обычно развиваются в позднем детстве или в раннем взрослом возрасте; некоторые варианты могут быстро прогрессировать и быть опасными для жизни, тогда как другие развиваются медленно
- окулофарингеальный MD — тип MD, который обычно не развивается до тех пор, пока человеку не исполнится 50-60 лет, и не влияет на ожидаемую продолжительность жизни
- Emery-Dreifuss MD — тип MD, развивающийся в детстве или в раннем взрослом возрасте; большинство людей с этим заболеванием доживают по крайней мере до среднего возраста
Подробнее о видах МД и диагностировании МД.
Кто страдает мышечной дистрофией?
В Великобритании около 70 000 человек имеют MD или родственное заболевание.
Duchenne MD — наиболее распространенный тип MD. В Великобритании ежегодно рождается около 100 мальчиков с диагнозом Дюшенна, а в Великобритании одновременно проживает около 2500 человек с этим заболеванием.
Миотонический MD является вторым по распространенности типом MD, которым страдает примерно 1 человек из 8000.
Facioscapulohumeral MD считается одним из каждых 20000 человек в Великобритании, что делает его третьим по распространенности MD.
Диагностика мышечной дистрофии
Для диагностики различных типов МД можно использовать множество различных методов. Возраст, в котором диагностируется состояние, будет варьироваться в зависимости от того, когда впервые начинают проявляться симптомы.
Диагностика включает некоторые или все следующие этапы:
- исследование любых симптомов
- , обсуждая семейную историю MD
- медицинский осмотр
- анализы крови
- электрические пробы нервов и мышц
- биопсия мышцы (при которой для исследования берут небольшой образец ткани)
Обратитесь к терапевту, если у вас или у вашего ребенка есть какие-либо симптомы MD.При необходимости ваш терапевт может направить вас в больницу для проведения дополнительных анализов.
Лечение мышечной дистрофии
От MD неизлечимо, но ряд методов лечения может помочь с физическими недостатками и проблемами, которые могут развиться. Сюда могут входить:
- помощь при передвижении, включая упражнения, физиотерапию и физические вспомогательные средства
- группы поддержки — чтобы справиться с практическим и эмоциональным воздействием MD
- хирургия — для исправления постуральных деформаций, таких как сколиоз
- лекарства, такие как стероиды для увеличения мышечной силы или ингибиторы АПФ и бета-блокаторы для лечения сердечных заболеваний
Новое исследование изучает способы исправления генетических мутаций и поврежденных мышц, связанных с MD.В настоящее время существуют многообещающие клинические испытания MD Duchenne.
Подробнее о лечении MD.
Информация о вас
Если у вас есть MD, ваша клиническая бригада передаст информацию о вас в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать более эффективные способы профилактики и лечения этого состояния. Вы можете отказаться от регистрации в любое время.
Узнайте больше о реестре.
Группы поддержки
MD может повлиять на вас как эмоционально, так и физически. Группы поддержки и организации могут помочь вам понять и смириться с вашим заболеванием. Они также могут предоставить полезные советы и поддержку тем, кто заботится о людях с МД.
Есть несколько национальных благотворительных организаций, которые предлагают поддержку людям, затронутым MD, например, Muscle Help Foundation и Muscular Dystrophy UK. Вы также можете спросить у своего терапевта или других лечащих вас медицинских работников о местных группах поддержки.
Есть также несколько национальных групп поддержки, которые продвигают исследования или оказывают поддержку конкретным типам MD, такие как Action Duchenne, Duchenne UK и Группа поддержки миотонической дистрофии.
Последняя проверка страницы: 24 мая 2018 г.
Срок следующей проверки: 24 мая 2021 г.