Трисомия 18: Синдром Эдвардса Статьи
Синдром Эдвардса – это редкое генетическое заболевание, связанное с дублированием 18 хромосомы. Впервые описано это заболевание было в 1980 году доктором Джоном Эдвардсом, который описал симптомы. Трисомия 18 может выражаться в полной и в мозаичной форме. При этом мозаичная форма синдрома имеет более благоприятный прогноз.
Диагностика синдрома
В связи с появлением современных технологий, сейчас возможно раннее прогнозирование синдрома Эдвардса – начиная с ранних сроков беременности. В случае, если установлен синдром, решение о сохранении беременности принимает женщина. В редких случаях генетическая патология не выявляется до момента появления ребенка на свет. После рождения ребенка трисомия 18 может устанавливаться по внешним признакам, однако точный ответ может дать только кариотипический анализ. Он же поможет определить, полная форма синдром у больного, или мозаичная. В случае подозрения на синдром Эдвардса, пациент нуждается в консультации генетика с последующей сдачей анализов.
Признаки трисомии 18
Первые признаки генетического отклонения у плода появляются еще во время беременности. Вот самые частые из них: сильная задержка внутриутробного развития на несколько недель, пороки развития головного мозга, многоводие, слабые шевеления плода, наличие только одной пупочной артерии. Подобная беременность часто длится 42 недели и более, а ребенок при этом рождается с небольшим весом.
Характерные для синдрома Эдвардса внешние признаки сразу заметны даже у новорожденного ребенка. Такой ребенок будет иметь низкую массу тела, долихоцефалический череп, низкий лоб и широкий затылок, недоразвитые и низкорасположенные ушные раковины. Небо у таких детей обычно высокое, «готическое», верхняя губа короче, чем у обычных детей. Может встречаться множество различных изменений опорно-двигательного аппарата: расширенная грудная клетка, узкий таз, вывихи бедер, стопы-«качалки». Пальцы на ногах часто сросшиеся или имеют перепонки. Пальцы рук скрещены и укорочены.
Часто встречаются проблемы с недоразвитием или неправильным развитием органов пищеварения и мочеполовой системы.
Такие дети обычно имеют проблемы с глотанием, сосанием. Нередки случаи рефлюкса и очень часто требуется зондовое выкармливание подобных детей. Многие дети с трисомией 18 предрасположены к возникновению судорог, а также – к необъяснимым скачкам температуры без других симптомов.
Вес у таких детей прибавляется очень плохо, нередко к году ребенок с синдромом Эдвардса не доходит до планки даже в 5 кг.
Умственное развитие детей с синдромом Эдвардса обычно соответствует тяжелым степеням олигофрении, однако дети с мозаичной формой синдрома иногда добиваются определенных успехов в умственном развитии.
Прогноз для людей с синдромом Эдвардса
Как и любое другое генетическое заболевание, синдром Эдвардса не лечится. Однако возможно симптоматическое лечение, связанное с облегчением жизни данному ребенку, в том числе и хирургическое лечение пороков, сопутствующих синдрому. Однако решение о целесообразности операции должны принимать врачи, поскольку зачастую риск осложнений превышает возможную пользу от операции.
Однако решение о целесообразности операции должны принимать врачи, поскольку зачастую риск осложнений превышает возможную пользу от операции.
Прогноз для жизни детей, у которых стоит диагноз «синдромом Эдвардса», в России в настоящее время неблагоприятен. Лишь небольшой процент детей с полной формой синдрома доживает до года. Средний срок жизни мальчиков ограничивается 2-3 месяцами, девочек – 10 месяцами. Однако при хорошем уходе со стороны родителей и внимательном наблюдении врачей дети с синдромом Эдвардса могут прожить гораздо дольше и иметь другое качество жизни. Они вполне могут научиться узнавать родных, улыбаться, самостоятельно кушать – при условии внимательного и заботливого к ним отношения. Дети с мозаичной формой синдрома имеют еще больше шансов на продолжительную и более полноценную жизнь.
В Европе и Америке люди с полной формой синдрома Эдвардса доживают до совершеннолетия. В России таких случаев пока зарегистрировано не было. Однако медицина не стоит на месте, и образованность людей повышается, поэтому вполне возможно, что скоро прогнозы для жизни людей с синдромом Эдвардса в России станут более благоприятными.
Синдром Эдвардса, ДНК-диагностика синдрома Эдвардса
Синдром Эдвардса (трисомия по хромосоме 18) — второе по частоте после синдрома Дауна хромосомное нарушение. Частота синдрома Эдвардса составляет 1:5000-1:7000 новорождённых. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков.
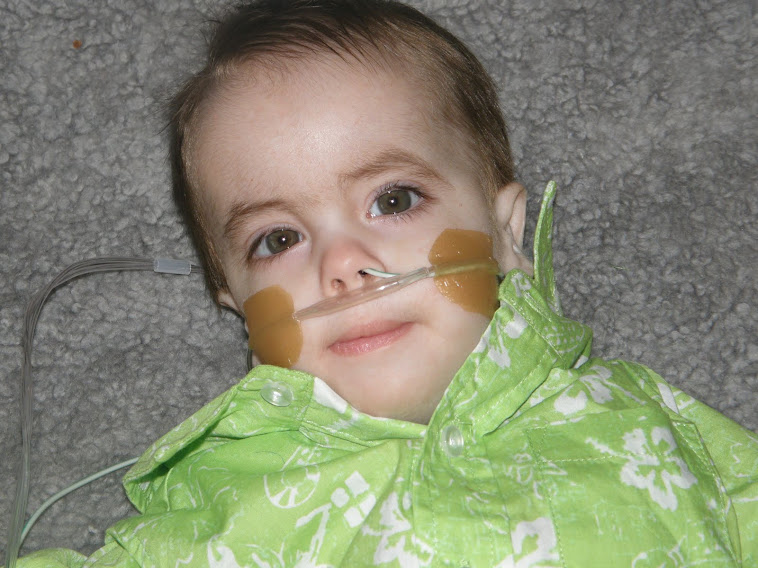
| Рисунок 1. Пример диагностики синдрома Эдвардса методом КФ-ПЦР. | Рисунок 2. Больной ребенок с синдромом Эдвардса. |
Пример диагностики синдрома Эдвардса (трисомии по хромосоме 18). Фиолетовым цветом выделены маркеры, расположенные в районе хромосомы 18, критическом для развития синдрома Эдвардса. В генотипе исследуемого образца по маркеру D18S978 наблюдается 1 пик (маркер неинформативен), по маркерам D18S535 и D18S386 — 3 пика (трисомия), по маркерам D18S390 и D18S819 — эффект дозы – неравное соотношение высоты двух пиков (трисомия). Таким образом, по четырем маркерам (D18S535, D18S386, D18S390 и D18S819) обнаружена трисомия, что позволяет установить диагноз «синдром Эдвардса». Генетический пол соответствует мужскому — присутствуют пики, соответствующие Y-хромосоме, по маркерам Amelogenin, 4SH, ZFXY, TAFL и присутствует пик гена SRY. Таким образом, по четырем маркерам (D18S535, D18S386, D18S390 и D18S819) обнаружена трисомия, что позволяет установить диагноз «синдром Эдвардса». Генетический пол соответствует мужскому — присутствуют пики, соответствующие Y-хромосоме, по маркерам Amelogenin, 4SH, ZFXY, TAFL и присутствует пик гена SRY. |
|
«Золотым стандартом» выявления хромосомных нарушений во всем мире долгое время являлся и продолжает оставаться метод кариотипирования с дифференциальной окраской хромосом. Этот метод позволяет анализировать кариотип в целом и определять крупные (не менее 5-10 млн пар нуклеотидов) хромосомные перестройки. Однако у него существует ряд ограничений, таких как трудоемкость, длительность (1-2 недели), высокие требования к квалификации и опыту специалиста, проводящего исследование, а также, в ряде случаев, технические проблемы (недостаточное количество и качество исследуемого материала, отсутствие митозов или роста культуры).
Этих недостатков лишен метод количественной флуоресцентной полимеразной цепной реакции (КФ-ПЦР), который все более широко применяется для диагностики анеуплоидий, в том числе и синдрома Эдвардса (Рис. 1). Этот метод обладает достоверностью, сравнимой с достоверностью стандартного кариотипирования, является более быстрым, дешевым, менее требовательным к количеству и качеству материала (поскольку не связан с ростом культуры клеток) и позволяет одновременно анализировать большое число образцов. Однако метод КФ-ПЦР имеет и ограничения: в мозаичных случаях он позволяет выявлять только высокоуровневый мозаицизм (от 20%), кроме того, он не может исключить наличие более редких хромосомных нарушений, которые могут быть связаны с пороками развития плода. При проведении дородовой диагностики синдрома Эдвардса, кроме материала плода, необходимо предоставлять биологический материал матери для того, чтобы исключить возможность получения ложноотрицательного результата из-за неправильного забора плодного материала.
При синдроме Эдвардса отмечается выраженная задержка пренатального развития, дети рождаются с пренатальной гипотрофией (средняя масса тела при рождении составляет 2340 г). Внешние проявления синдрома Эдвардса многообразны (Рис. 2). Наиболее типичными являются задержка психомоторного развития, гипоплазия скелетной мускулатуры и подкожной жировой ткани, врожденные пороки сердца, аномалии строения лица и черепа (долихоцефалия, микрофтальмия, укорочение глазных щелей, низкое расположение ушных раковин, микрогнатия, скошенный подбородок), множественные деформации кистей и стоп, аномалии развития желудочно-кишечного тракта, мочеполовой системы и центральной нервной системы (спинно-мозговые грыжи, гипоплазия мозолистого тела и мозжечка). Продолжительность жизни детей резко снижена: 90% из них погибают до года от осложнений, обусловленных врождёнными пороками развития (асфиксия, пневмония, кишечная непроходимость, сердечно-сосудистая недостаточность).
Причиной развития синдрома Эдвардса является утроение хромосомы 18. Трисомия по хромосоме 18 является частным случаем анеуплоидии– наличия в геноме набора хромосом, отличного от стандартного для данного вида и некратного ему. Трисомия хромосомы 18 обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (яйцеклеток и сперматозоидов), в результате чего ребенок получает от матери или от отца лишнюю 18-ю хромосому. В этом случае все клетки организма ребёнка будут нести аномалию. В том случае, когда нерасхождение хромосом возникает при делении какой-либо клетки зародыша, наблюдается мозаичный вариант синдрома Эдвардса (10% случаев).
Риск рождения детей с синдромом Эдвардса, по разным литературным данным, не изменяется или незначительно возрастает с увеличением возраста беременной женщины.
Пренатальная диагностика синдрома Эдвардса включает в себя два этапа. На первом этапе, на сроке беременности 11-13 недель, проводится скрининг, который основывается преимущественно на биохимических показателях, поскольку на ранних сроках УЗИ не позволяет обнаружить в случае синдрома Эдвардса каких-либо грубых аномалий развития, которые могут быть выявлены лишь к 20-24 неделе.
В Центре Молекулярной Генетики проводится диагностика синдрома Эдвардса (в том числе и пренатальная) методом КФ-ПЦР.
Синдром Эдвардса
Трисомия 18 (синдром Эдвардса) является наиболее распространенной аутосомной аномалией среди родившихся живыми детей после синдрома Дауна (трисомия 21). В большинстве случаев трисомия 18 (синдром Эдвардса) является истинной, т.е. ее причиной является нерасхождение хромосом во время мейоза. В небольшом числе случаев (до 10%) трисомия 18 (синдром Эдвардса) является мозаичной (мозаицизм — состояние, при котором в организме присутствуют ткани генетически различных типов, например, нормальные клетки и клетки имеющие три хромосомы), вызванные постзиготным нерасхождением хромосом в анафазе — на ранней стадии развития зародыша (Chen 2004, Forrester 1999, Carothers 1999, Huether 1996, Pradat 1991, Buyse, 1990. Возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется «Транслокация», и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша.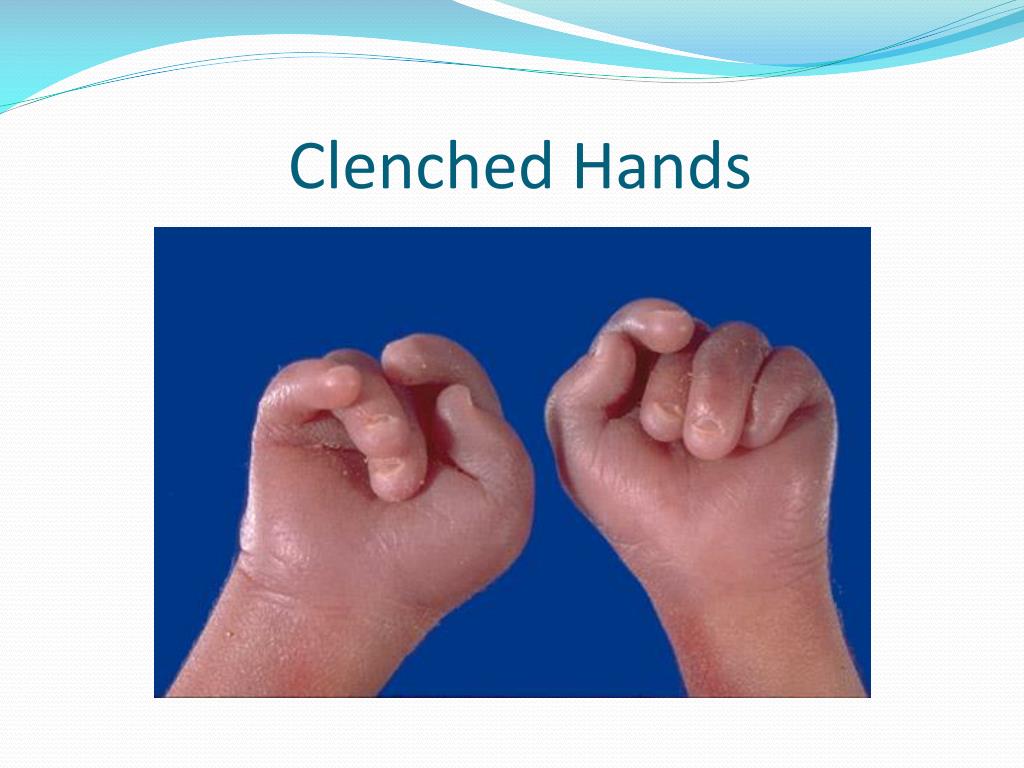 В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикрепленная к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса. Большинство трисомий 18 обнаруживаются у плодов в середине второго триместра и очень часто такие беременности заканчиваются внутриутробной гибелью плода. (Hook, 1989).
В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикрепленная к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса. Большинство трисомий 18 обнаруживаются у плодов в середине второго триместра и очень часто такие беременности заканчиваются внутриутробной гибелью плода. (Hook, 1989).
Клинические особенности, связанные с трисомии 18 включают, но не ограничиваются следующими: расстройства центральной нервной системы (голопрозенцефалия, менингомиелоцеле), пороки развития глаз, носа, расщелина губы и / или неба, деформация ушных раковин, деформированные конечностей, полидактилия, а также дефекты сердца, половых органов, и пр.
Прогноз для этого заболевания, как правило, неблагоприятный. Большинство рожденных детей, умирают, в среднем, в течение 2 — 10 дней (Parker, 2003). Тем не менее, есть дети, продолжительность жизни которых достигает года и более (Rasmussen, 2003).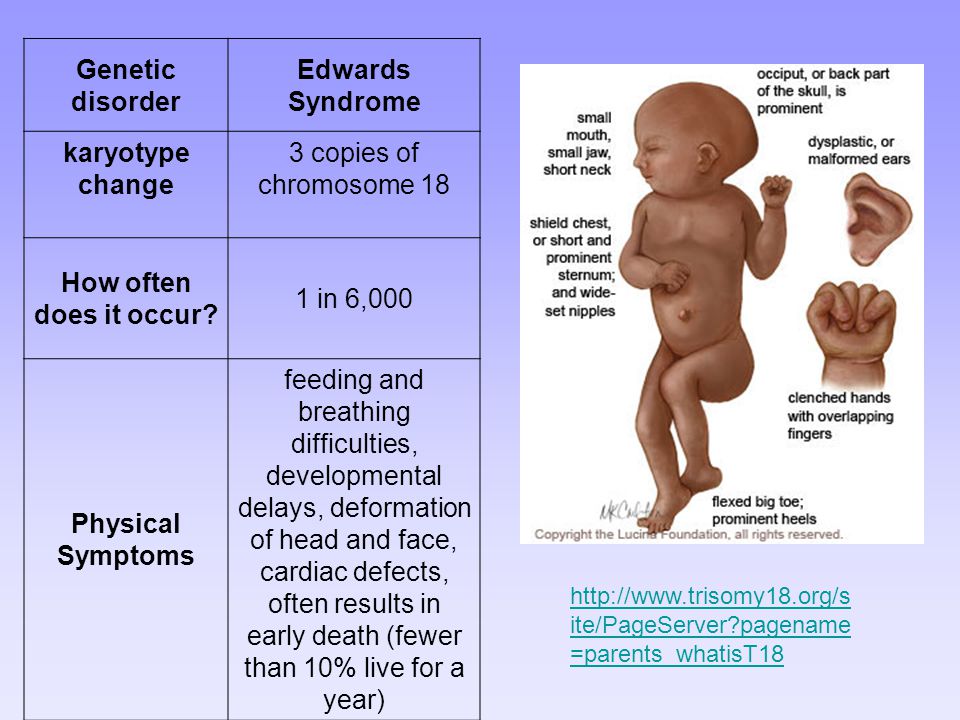 Однако, такие дети имеют как физическое, так и психическое отставание в развитии (Parter 2003). Популяционная частота примерно 1:7000.
Однако, такие дети имеют как физическое, так и психическое отставание в развитии (Parter 2003). Популяционная частота примерно 1:7000.
ЭТИОЛОГИЯ
Причиной истинной трисомии 18 является нерасхождение хромосом при формировании яйцеклетки и сперматозоида, когда одна гамета получает дополнительную 18 хромосому. Нерасхождение может произойти в первом (MI) или втором (MII) мейотического этапе.
В 90-97% случаев дополнительная хромосома 18 имеет материнское происхождение, а отцовское происхождение в 3-10% всех случаев. Среди случаев трисомии 18 материнского происхождения, 31-39% бывают в результате нерасхождения хромосом в фазе MI и 61-69% в результате нерасхождение в фазе MII (Bugge et al., 1998; Nicolaidis and Petersen, 1998; Eggermann et al., 1996; Ramesh and Verma, 1996; Fisher et al., 1995; Jacobs and Hassold, 1995; Fisher et al., 1993; Ya-gang et al., 1993).
ДЕМОГРАФИЯ И РЕПРОДУКТИВНОСТЬ
Риск трисомии 18, как известно, возрастает с увеличением возраста матери (Munne 2004, Naguib 1999, Baty 1994, Buyse 1990, Goldstein 1988, Schreinemachers 1982).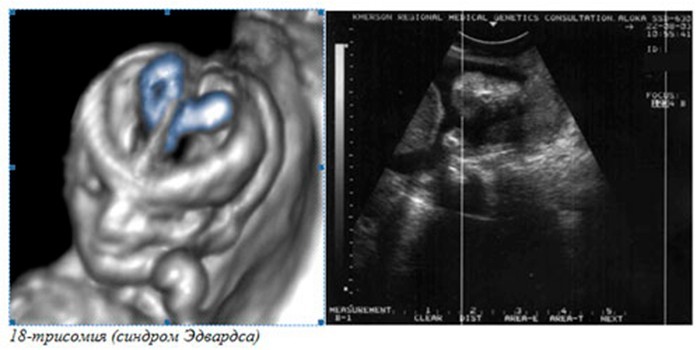 Риск трисомии 18 (синдрома Эдвардса) связывают также и с увеличением отцовского возраста, однако, если риск трисомии 18 увеличивается за счет возраста матери, то возрастом отца в таких случаях пренебрегают. (Naguib 1999, Baty 1994). Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %.
Риск трисомии 18 (синдрома Эдвардса) связывают также и с увеличением отцовского возраста, однако, если риск трисомии 18 увеличивается за счет возраста матери, то возрастом отца в таких случаях пренебрегают. (Naguib 1999, Baty 1994). Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %.
Раса/этническая принадлежность практически не влияет на риск трисомии 18 (Buyse 1990). Одно исследование показало, что из четырех расовых/этнических групп обследованных (европейцы, дальневосточные азиаты, жители островов тихого океана, филиппинцы), риск трисомии 18 был самым высоким для дальневосточных азиатов и самым низким для жителей островов тихого океана (Forrester, 1999). Тем не менее, эти различия в риске по видимому, связаны с различиями в распределении материнского возраста среди этих расовых / этнических групп.
Географическая область может влиять на риск трисомии 18. В одном исследовании приводятся данные, что риск трисомии 18 выше среди городских жителей (Forrester, 1999). Этот повышенный риск остается после учета возраста матери. Несколько исследований показали, что на распространенность трисомии 18 могут влиять сезонные колебания (Naguib, 1999).
Этот повышенный риск остается после учета возраста матери. Несколько исследований показали, что на распространенность трисомии 18 могут влиять сезонные колебания (Naguib, 1999).
Некоторые исследования показали, что Трисомия 18 (синдром Эдвардса) в последние годы имеет тенденцию к увеличению. Однако, возможно, это связано как с улучшением диагностики анеуплоидий, в т.ч. пренатальной диагностики и диагностики фетусов (Pradat 1991), так и с повышением возраста рожающих женщин.(Gessner 2003, Forrester, 1999).
За последние несколько десятилетий, было установлено, что в сыворотке женщин с плодом с трисомией 18 имеется низкий уровень альфа-фетопротеина, хорионический гонадотропин человека, и эстриол (anick 1993, Greenberg 1992, Doran 1986). Кроме того, при пренатальном ультразвуковом исследовании обнаруживаются различные структурные аномалии часто связаны с трисомией 18 (Abramsky 1993, Vintzileos 1987). Пренатальный скрининг маркеров, УЗИ и окончательный диагноз подтвержденный кариотипированием с помощью таких процедур, как амниоцентез и биопсия хориона, позволяют определить трисомию 18 в период внутриутробного развития.
Пол ребенка влияет на риск трисомии 18. Девочки с трисомией 18 рождаются в 3 раза чаще мальчиков. (Forrester 1999, Naguib 1999, Carothers 1999, Huether 1996, Pradat 1991, Buyse 1990, Goldstein 1988). Одно исследование показало, что соотношение полов изменяется в различных расовых /этнических группах, однако это такие наблюдения сделаны на небольших выборках, что не позволяет говорить о такой связи достоверно.(Huether 1996).
Риск повторения для трисомии 18 составляет, примерно, 1% (Baty 1994, Buyse 1990). Последние исследования показали, что риск трисомии увеличивается у женщин, которые имели трисомии 18 в предыдущих беременностях, независимо от рождения живого ребенка или внутриутробной гибели плода. То есть, даже если беременность самопроизвольно прерывается, риск остается повышенным (Munne 2004). Существует также повышенный риск трисомной беременности у женщин, с низким числом овоцитов (Kline, 2000). Вероятно, это условие связано с наступлением менопаузы.
ОБРАЗ ЖИЗНИ И ФАКТОРЫ ОКРУЖАЮЩЕЙ СРЕДЫ.

Нет достоверных данных, которые подтверждали бы влияние факторов окружающей среды на риск трисомии 18. Тем не менее, различия в распространенности трисомии 18 между популяциями (Forrester 1999, Naguib 1999) позволяют предположить, что экологические факторы могут влиять на риск хромосомных ошибок. В то же время, отсутствует повышенный риск трисомии 18 у людей живущих рядом со свалками твердых отходов или мусоросжигательными заводами (Cordier 2004, Harrison 2003). Наличие хлоридов и нитратов в питьевой воде (Cedergren 2002) , а также пестицидов (Berkowitz 2003) не увеличивают риск этого дефекта. У матерей, чья работа связана с химическими растворителями, также не увеличивает риск трисомии 18 (Wennborg 2005).
Вероятность хромосомных ошибок в настоящее время может увеличиваться из-за вспомогательных репродуктивных технологий (ВРТ). Однако, до конца не ясно, связано ли это с несовершенством лабораторных методов или с генетическими нарушениями у родителей, которые одновременно могут являться и причиной бесплодия.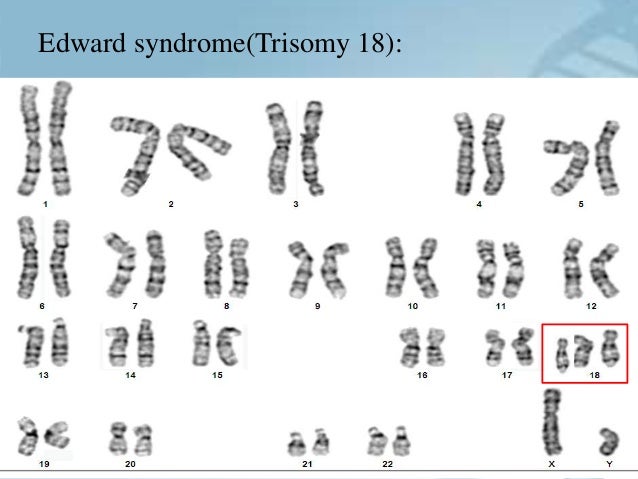 То есть пары, которые не могут забеременеть естественным путем могут быть предрасположены к генетическим ошибкам (Palermo 2000).
То есть пары, которые не могут забеременеть естественным путем могут быть предрасположены к генетическим ошибкам (Palermo 2000).
Одно исследование показало, что различия в метаболизме фолиевой кислоты у женщин не связаны с мейотическими ошибками приводящими к анеуплоидиям.
Регулярное употребление поливитаминов не связано с уменьшением риска трисомии 18 (Botto 2004).
ГЕНЕТИЧЕСКАЯ ПРЕДРАСПОЛОЖЕННОСТЬ
Иногда, вследствие явления, которое называется сбалансированная транслокация, некоторые люди могут нести генетический материал, принадлежащий хромосоме 18 в другой хромосоме. Поскольку у таких людей нет дополнительного генетического материала они не имеют признаков трисомии 18. Однако, у таких людей есть повышенный риск рождения детей с этим заболеванием. Установить носительство сбалансированной хромосомной транслокации можно при исследовании кариотипа.
В редких случаях один из родителей может быть носителем частичной трисомии 18, которая может передаваться по наследству.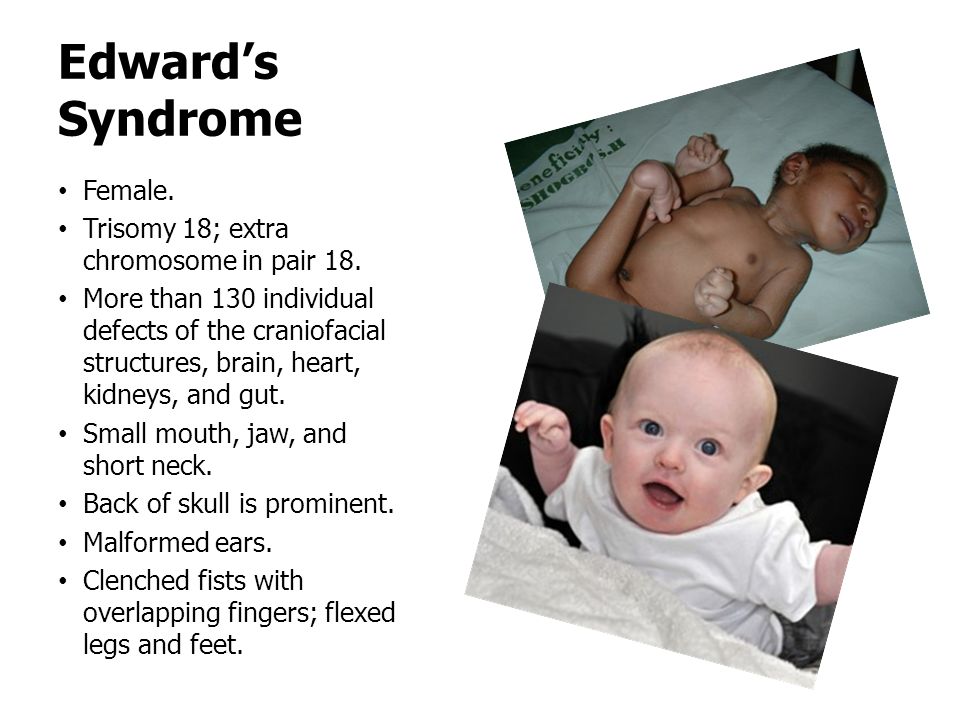 Установить носительство частичной трисомии можно с помощью хромосомного микроматричного анализа.
Установить носительство частичной трисомии можно с помощью хромосомного микроматричного анализа.
Таким образом, в большинстве случаев синдром Эдвардса является результатом случайных ошибок в делении клетки и мало связан с какими либо факторами окружающей среды или состояния человека.
Литература
- Abramsky L, Chapple J. Room for improvement? Detecting autosomal trisomies without serum screening. Public Health 1993;107:349-354.
- Baty BJ, Blackburn BL, Carey JC. Natural history of trisomy 18 and trisomy 13. I. Growth, physical assessment, medical histories, survival, and recurrence risk. Am J Med Genet 1994;49:175-188.
- Berkowitz GS, Obel J, Deych E, Lapinski R, Godbold J, Liu Z, Landrigan PJ, Wolff MS. Exposure to indoor pesticides during pregnancy in a multiethnic urban cohort. Environmental Health Perspectives 2003;111:1:79-84.
- Botto LD, Mulinare J, Yang Q, Liu Y, Erickson JD. Autosomal trisomy and maternal use of multivitamin supplements.
 American Journal of Medical Genetics 2004:125A:113-116.
American Journal of Medical Genetics 2004:125A:113-116. - Buyse ML, editor-in-chief. Birth Defect Encyclopedia . Cambridge, Massachusetts: Blackwell Scientific Publications, 1990.
- Canfield MA, Honein MA, Yuskiv N, Xing J, Mai CT, Collins JS, Devine O, Petrini J, Ramadhani TA, Hobbs CA, Kirby RS. National estimates and race/ethnic-specific variation of selected birth defects in the United States, 1999-2001. Birth Defects Res A Clin Mol Teratol. 2006 Nov;76(11):747-56.
- Canick JA, Saller DN. Maternal serum screening for aneuploidy and open fetal defects. Obstet Gynecol Clin North Am 1993;20:443-454.
- Cardonick E, Iacobucci A. Use of chemotherapy during human pregnancy. The Lancet Oncology 2004;5:283-291.
- Carothers AD, Boyd E, Lowther G, Ellis PM, Couzin DA, Faed MJ, Robb A. Trends in prenatal diagnosis of Down syndrome and other autosomal trisomies in Scotland 1990 and 1994, with associated cytogenetic and epidemiological findings. Genet Epidemiol 1999;16:179-190.
- Cedergren MI, Selbing AJ, Lofman O, Kallen BAJ. Chlorination byproducts and nitrate in drinking water and risk for congenital cardiac defects. Environmental Research Section A 2002;89:124-130.
- Chen M, Yeh GP, Shih JC, Wang BT. Trisomy 13 mosiacism: study of serial cytogentic changes in a case from early pregnancy to infancy. Prenatal Diagnosis 2004;24:137-143.
- Cordier S, Chevrier C, Robert-Gnansia E, Lorente C, Brula P, Hours M. Risk of congenital anomalies in the vicinity of municipal solid waste incinerators. Occup Environ Med 2004;61:8-15.
- Doran TA, Cadesky K, Wong PY, Mastrogiacomo C, Capella T. Maternal serum alpha-fetoprotein and fetal autosomal trisomies. Am J Obstet Gynecol 1986;154:277-281.
- Forrester MB, Merz RD. Trisomies 13 and 18: Prenatal diagnosis and epidemiologic studies in Hawaii, 1986-1997. Genet Test 1999;3:335-340.
- Forrester MB , Merz RD, Yoon PW. Impact of prenatal diagnosis and elective termination on the prevalence of selected birth defects in Hawaii. Am J Epidemiol 1998;148:1206-1211.
- Fried PA. The consequences of marijuana use during pregnancy: a review of the human literature, in Women and Cannabis: Medicine, Science, and Sociology, The Haworth Integrative Healing Press, 2002.
- Gessner BD. Reasons for trisomy 13 and 18 births despite the availability of prenatal diagnosis and pregnancy termination. Early Human Development 2003; 73:53-60.
- Goldstein H, Nielsen KG. Rates and survival of individuals with trisomy 13 and 18. Clin Genet 1988;34:366-372.
- Greenberg F, Schmidt D, Darnule AT, Weyland BR, Rose Esmie, Alpert E. Maternal serum alpha-fetoprotein, beta-human chorionic gonadotropin, and unconjugated estriol levels in midtrimester trisomy 18 pregnancies. Am J Obstet Gynecol 1992;166:1388-1392.
- Harrison RM. Hazardous waste landfill sites and congenital anomalies. Occup Environ Med 2003; 60:79-80.
- Hassold TJ, Burrage LC, Chan ER, Judis LM, Schwartz S, James SJ, Jacobs PA, Thomas NS. Maternal folate polymorphisms and the etiology of human nondisjuntion. American Journal of Human Genetics; 2001:69:434-439.
- Hook EB, Topol BB, Cross PK. The natural history of cytogenetically abnormal fetuses detected at midtrimester amniocentesis that are not terminated electively: New data and estimates of the excess and relative risk of late fetal death associated with 47,+21 and other abnormal karyotypes. Am J Hum Genet 1989;45:855-861.
- Huether CA, Martin LM, Stoppelman SM, D’Souza S, Bishop JK, Torfs CP, Lorey F, May KM, Hanna JS, Baird PA, Kelly JC. Sex ratios in fetuses and liveborn infants with autosomal aneuploidy. Am J Med Genet 1996;63:492-500.
- Jeng W, Wong AW, Ting-A-Kee R, Wells PG Methanmphetamine-enhanced embryonic oxidative DNA damage and neurodevelopmental deficits.Free Radical Biology and Medicine 2005; 39:317-326.
- Kallen B. Use of antihistamine drugs in early pregnancy and delivery outcome. Journal of Maternal-Fetal and Neonatal Medicine 2002;11:146-152.
- Kline J, Kinney A, Levin B, Warburton D. Trisomic pregnancy and earlier age at menopause. American Journal of Human Genetics 2000;67:395-404.
- Kupke KG, Muller U. Parental origin of the extra chromosome in trisomy 18. Am J Hum Genet 1989;45:599-605.
- Munne S, Sandilinas M, Magli C, Gianaroli L, Cohen J, Warburton D. Increased rate of aneuploid embryos in young women with previous aneuploid conceptions. Prenatal Diagnosis 2004; 24: 638-643.
- Naguib KK, Al‑Awadi SA, Moussa MA, Bastaki L, Gouda S, Redha MA, Mustafa F, Tayel SM, Abulhassan SA, Murthy DS. Trisomy 18 in Kuwait. Int J Epidemiol 1999;28:711-716.
- Nothen MM, Eggermann T, Erdmann J, Eiben B, Hofmann D, Propping P, Schwanitz G. Retrospective study of the parental origin of the extra chromosome in trisomy 18 (Edwards syndrome). Hum Genet 1993;92:347-349.
- Palermo GD, Neri QV, Hariprashad JJ, Davis OK, Veeck LL, Rosenwaks Z. ICSI and its outcome. Seminars in Reproductive Medicine 2000; 18:2: 161-169.
- Parker MJ, Budd JLS, Draper ES, Young ID. Trisomy 13 and trisomy 18 in a defined population: epidemiological, genetic, and prenatal observations. Prenatal Diagnosis 2003;23:856-860.
- Park-Wyllie L, Mazzotta P, Pastuszak A, Moretti ME, Beique L, Hunnisett L, Friesen MH, Jacobson S, Kasapinovic S, Chang D, Diav-Citrin O, Chitayat D, Nulman I, Einarson TR, Koren G. Birth defects after maternal exposure to corticosteroids: prospective cohort study and meta-analysis of epidemiological studies. Teratology 2000;62:385-392.
- Pradat P. Is trisomy 18 increasing in Sweden? An analysis of the syndrome during a ten-year period and a comparison with a French registry. Hereditas 1991;114:97-102.
- Ramesh KH, Verma RS. Parental origin of the extra chromosome 18 in Edwards syndrome. Ann Genet 1996;39:110-112.
- Rasmussen SA, Wong LYC, Yang Q, May KM, Friedman JM. Population based analysis of mortality in trisomy 13 and trisomy 18. Pediatrics 2003;111:777-784.
- Schreinemachers DM, Cross PK, Hook EB. Rates of 47,+21, 47,+18, 47,+13 and other chromosome abnormalities in about 20,000 prenatal studies compared with estimated rates in live births. Hum Genet 1982;61:318-324.
- Sorensen HT, Czeizel AE, Rockenbauer M, Steffensen FH, Olsen J. The risk of limb deficiencies and other congenital abnormalities in children exposed to calcium channel blockers. Acta Obst Gynecol Scand 2001;80:397-401.
- Vintzileos AM, Campbell WA, Nochimson DJ, Weinbaum PJ. Antenatal evaluation and management of ultrasonically detected fetal anomalies. Obstet Gynecol 1987;69:640-660.
- Wennborg H, Magnusson LL, Bonde JP, Olsen J. Congenital malformations related to maternal exposure to specific agents in biomedical research laboratories. JOEM 2005; 47:1:11-19.
- Vrijheid M, Dolk H, Stone D, Abramsky L, Alberman E, Scott JES. Socioeconomic inequalities in the risk of congenital anomaly. Arch Dis Child 2000; 82:349-352.
- Ya-gang X, Robinson WP, Spiegel R, Binkert F, Ruefenacht U, Schinzel AA. Parental origin of the supernumerary chromosome in trisomy 18. Clin Genet 1993;44:57-61.
 American Journal of Medical Genetics 2004:125A:113-116.
American Journal of Medical Genetics 2004:125A:113-116. Genet Epidemiol 1999;16:179-190.
Genet Epidemiol 1999;16:179-190. Impact of prenatal diagnosis and elective termination on the prevalence of selected birth defects in Hawaii. Am J Epidemiol 1998;148:1206-1211.
Impact of prenatal diagnosis and elective termination on the prevalence of selected birth defects in Hawaii. Am J Epidemiol 1998;148:1206-1211. Occup Environ Med 2003; 60:79-80.
Occup Environ Med 2003; 60:79-80.
 Hum Genet 1993;92:347-349.
Hum Genet 1993;92:347-349.Трисомия 18 (синдром Эдвардса) ᐉ диагностика, лечение в МЦ БОГОЛЮБЫ
Трисомия 18 — генетическое нарушение, известное также как синдром Эдвардса, характеризующееся набором хромосомных изменений, которые могут быть причиной внутриутробной смерти плода или гибели ребенка в ближайшее время после рождения.
Диагностировать трисомию можно на ранних сроках, благодаря скрининговым тестам, проведенным во время беременности. Но, к сожалению, на сегодняшний день заболевание является неизлечимым. Смерть наступает из-за серьезных поражений сердца, почек, легких, желудочно-кишечного тракта и нервной системы.
Прежде чем говорить о сути заболевания, необходимо немного освежить в памяти наши познания в генетике. Итак, каждая клетка здорового человека имеет 23 пары хромосом. Одна пара из этих хромосом — половая, то есть, определяет пол человека, остальные 22 — аутосомные хромосомы. В своей совокупности, эти 46 хромосом содержат весь генетический материал человека, более известный как ДНК. В ДНК индивидуума записано все о нем — черты лица, физические особенности, предрасположенности к болезням и т.п.
Каждая пара хромосом содержит определенные гены. Когда в хромосоме происходит мутация, какой-то ген может оказаться «неисправным», и соответственно дефектный ген начинает производить дефектный белок. Когда изменения касаются количественных характеристик хромосом, говорят об анеуплоидии. В этом случае вместо двух хромосом в паре может быть три (трисомии) или одна (моносомии).
Синдром Эдвардса, или трисомия 18 — это генетическое нарушение, характеризующееся наличием третьей хромосомы 18. Таким образом, хромосомный набор человека состоит из 47 хромосом, вместо 46. У синдрома Эдвардса нет благоприятного прогноза, как правило, ребенок умирает в течение первого года жизни.
Трисомия 18 является вторым по распространенности генетическим нарушением после трисомии 21 (синдрома Дауна), его частота составляет один случай на 6000 живорожденных. Нужно учесть, что в большинстве случаев синдром Эдвардса связан с внутриутробной смертью. Выживают, как правило, девочки.
Причины трисомии 18
Что же становится причиной трисомии 18? На этот вопрос до сих пор нет вразумительного ответа. Возможно, происходит какая-то генетическая ошибка во время мейоза половых клеток, или митоза оподотворенной яйцеклетки, или эмбриона.
Факторы риска трисомии 18
Случаи трисомии 18 характерны для беременных в возрасте старше 40 лет. Но это не единственный фактор риска. Семейный анамнез также имеет значение. Если в семейной истории были случаи трисомии 18, то велика вероятность повторения заболевание в поколениях.
Симптомы трисомии 18
В предродовой период трисомию 18 отличают следующие признаки:
- многоводие;
- маловодие;
- задержка внутриутробного развития;
- слабая активность плода.
Основной причиной внутриутробной смерти плода при трисомии 18 являются сердечные патологии. Младенцы с трисомией 18 имееют длинный ряд симптомов, затрагивающий множество органов и систем. Это скелетные, и сердечные аномалии, желудочно-кишечные, мочеполовые, неврологические и дыхательные нарушения. Физические характеристики черепа и лица также имеют свои особенности: уродливые уши, выступающая затылочная область черепа, маленький рот, маленькие глаза.
Диагностика трисомии 18
Обнаружить трисомию 18 можно с помощью амниоцентеза. Если генетическа аномалия имеет место, то при данном виде диагностике, она обнаруживается с высокой точностью.
Терапия трисомии 18
К сожалению, генетические ошибки мы пока еще не научились исправлять. Прогноз трисомии всегда неблагоприятный. Статистика неумолима: более 50% детей умирают в первую неделю жизни, 40% — доживают до месяца, 5% — до года, 1% — до 10 лет. Смерть у пациентов с трисомией 18 наступает из-за пороков развития сердца, почек, центральной нервной системы и опухолей печени и почек.
Важность определения хромосомных аномалий
Почему надо проводить тестированиена синдром Дауна?
Многие пары находят, что это помогает, если они знают до родов, что у их ребёнка синдром Дауна. Дети с синдромом Дауна нуждаются в дополнительной врачебной помощи в зависимости от проблем со здоровьем конкретного ребёнка.
Большинство людей с синдромом Дауна живёт до 60х годов, и многие даже дольше. Наличие проблем со здоровьем, например, пороков сердца, может повлиять на продолжительность жизни этих детей и взрослых. Около 30% беременностей с синдромом Дауна прерывается.
У большинства детей с синдромом Дауна дефект интеллекта, варьирующийся от лёгкого до умеренного. Показано, что раннее вмешательство может быть благоприятным для детей с синдромом Дауна. Многие дети с синдромом Дауна учатся читать и писать и участвуют в обычной деятельности.
У родителей, которые до родов узнают, что у их ребёнка синдром Дауна, есть возможность сделать эхокардиограмму плода, чтобы узнать, есть ли у их ребёнка нуждающийся в хирургическом вмешательстве после родов порок сердца. Если их ребёнку с синдромом Дауна требуется какая-либо операция, то до родов у родителей есть возможность встретиться с детским врачом, чтобы лучше подготовиться к операции, которая может понадобиться их ребёнку. В случае, если ребёнку требуется специальный уход сразу после родов, пара может в некоторых случаях выбрать для родов больницу, где есть отделение интенсивной терапии новорождённых высокого уровня. У пар также есть возможность изучить информацию о группах поддержки родителей детей с синдромом Дауна.Также пары могут пожелать узнать о возможностях раннего лечения синдрома Дауна.
Почему надо проводить тестированиена синдром Эдвардса ?
Трисомия 18, которая также называется синдромом Эрвардса, обусловлена дополнительной хромосомой 18 хромосомы. Примерно один ребёнок из 3000 рождается с этим заболеванием. К сожалению, большинство грудничков с трисомией 18 умирает в первые недели жизни, и менее чем 10% живут дольше 1 года.У грудничков с синдромом Эдвардса много дефектов интеллекта и пороков развития внутренних органов, которые обычно затрагивают сердце, мозг и почки, а также наружные пороки развития, как, например, заячья губа и волчья пасть, маленькая голова, косолапость, неразвитые пальцы рук и маленькая нижняя челюсть. У беременностей с диагнозом трисомии 18 большой риск прерваться или закончиться мертворождением. Трисомия 18 появляется случайно. Во время беременности Вы не можете ничего сделать для предотвращения трисомии 18.
Почему надо проводить тестированиена синдром Патау ?
Трисомия 13, которая также называется синдромом Патау, обусловлена дополнительной хромосомой 13 хромосомы. Примерно один ребёнок из 5000 рождается с трисомией 13. Дети с трисомией 13 нуждаются в значительной врачебной помощи, потому что у них много дефектов интеллекта и врождённых дефектов. У них могут быть врождённые дефекты, проблемы с головным или спинным мозгом, дополнительные пальцы рук и/или ног, волчья пасть или с, или без заячьей губы, и слабый мышечный тонус. У многих детей также есть врождённые дефекты других органов. Большинство детей с трисомией 13 не живёт дольше одной недели. У беременностей с диагнозом трисомии 13 большой риск прерваться или закончиться мертворождением. Трисомия 13 появляется случайно. Во время беременности Вы не можете ничего сделать для предотвращения трисомии 13.
Почему надо проводить тестированиена синдромТёрнера?
Моносомия X (45,X), которая также называется синдромом Тёрнера- это хромосомное заболевание, причиной которого является отсутствие второй X-хромосомы у девочки. У человека обычно 46 хромосом, представленных 23 парами. У девочек обычно две X-хромосомы и у мальчиков одна X-хромосома и одна Y-хромосома. У девочек с моносомией X одна X-хромосома вместо двух.
В большинстве случаев девочки с моносомией Xнуждаются в дополнительной врачебной помощи. Эта дополнительная врачебная помощь может продолжаться в детском и подростковом возрасте, когда желательно использовать разное гормональное лечение. Родители, которые перед родами узнают, что у их ребёнка синдром Тёрнера, могут ходатайствовать о проведении детального ультразвукового исследования (эхокардиографии) сердца плода примерно на 20 неделе беременности. Если обнаружится врождённый порок сердца, родители могут встретиться с детскими кардиологами и изучить, что ждёт их ребёнка после рождения с точки зрения здоровья сердца. Родители также смогут больше узнать о синдроме Тёрнера и связаться с государственными и местными группами поддержки пациентов до рождения своего ребёнка.
Большая часть беременностей с моносомией X, то есть синдромом Тёрнера, прерывается в первом или втором триместре.Знание этого помогает парам лучше эмоционально приспособиться к этой возможности.
Почему надо проводить тестирование нааномалии половых хромосом?
Комбинированные аномалии половых хромосом чаще, чем синдром Дауна.
В случае детей с пренатально диагностированными аномалиями половых хромосом показано, что у них более лёгкий ход развития, чем у тех детей, чей диагноз был установлен после родов. Доступными вариантами лечения являются:
- рече-, физио- и/или трудотерапия;
- раннее образовательное вмешательство;
- гормональное лечение.
Капли материнской крови достаточно, чтобы узнать все о будущем ребенке – Наука – Коммерсантъ
текст Катерина Пантюх аспирант кафедры биотехнологии МГУ им. Ломоносова
Николай Чеканов научный сотрудник ЗАО «Геноаналитика»
иллюстрация Хадия Улумбекова
маргиналия Анна Кольцова
Среди всевозможных генетических нарушений особое место занимают хромосомные анеуплоидии (нарушения числа хромосом). Такие отклонения, как синдром Эдвардса, синдром Патау и синдром Дауна отличаются тяжелыми физическими нарушениями, умственными отклонениями и при этом имеют достаточно большую частоту встречаемости. Анеуплоидии никак не наследуются, их возникновение не зависит от образа жизни матери, социального статуса — только от возраста родителей.
В современной стандартной медицинской практике в России принята двухступенчатая система скрининга беременных на наличие хромосомных анеуплоидий. На первом этапе проводится неинвазивное исследование: ультразвуковое обследование и биохимический анализ крови. Полученные косвенные данные не очень точны и позволяют лишь сформировать группу риска беременных с повышенной вероятностью проблем у плода. Всем попавшим в группу риска по показаниям неинвазивного исследования, а также просто тем, кому 35 лет или больше, предлагается пройти инвазивное исследование: забор образца ткани плаценты или амниотической жидкости и цитогенетический анализ его хромосомного набора — кариотипирование. Этот метод обладает очень высокой степенью достоверности, однако чреват осложнениями и даже незапланированным прерыванием беременности.
Можно ли обойтись малой кровью? Оказалось, что да, причем буквально. Небольшие фрагменты плодного генома проходят через плацентарный барьер и попадают в кровоток матери. Внеклеточная ДНК (вкДНК) была обнаружена в 1948 году французскими учеными Манделем и Метэ, еще до открытия знаменитой двойной спирали и окончательного подтверждения, что именно ДНК является носителем генетической информации. ВкДНК представляет собой остатки ядерного материала разрушенных клеток и присутствует в норме даже у здоровых людей, например, как результат перестроек мышечной и жировой ткани после занятий спортом; значительное повышение ее концентрации может свидетельствовать о развитии тяжелой болезни воспалительного, аутоиммунного или онкологического характера. Полвека спустя группа оксфордских исследователей показала наличие плодной вкДНК у беременных: она появляется в результате естественного распада клеток трофобласта, плаценты и гемопоэтических клеток. Появление плодной вкДНК в крови матери заметно после пятой недели беременности, со временем ее количество увеличивается и к десятой неделе составляет в среднем 10-15%. С момента открытия предлагалось использовать этот феномен для безопасной проверки общих генетических характеристик плода. Практически сразу были разработаны неинвазивные методики определения резус-фактора и пола будущего ребенка, однако более тонкие материи, вроде подсчета числа хромосом, классическим методам молекулярной биологии оказались не под силу. На помощь пришли технологии массового параллельного секвенирования (чтения) ДНК, которые бурно развивались с конца 2000-х годов.
Российские генетики из компании «Геноаналитика» разработали так называемый «ДОТ-тест»: систему диагностики основных трисомий плода по материнской крови. За 2014 год по ней было проанализировано более тысячи образцов из двенадцати регионов РФ, а полученные показатели чувствительности (99,8%) и специфичности (99,6%) не уступают иностранным конкурентам. Суть методики проста. У беременной женщины берут небольшое количество венозной крови, из которой оставляют только плазму, выделяют ДНК и затем секвенируют. Геном плода представлен в вкДНК более или менее равномерно, соответственно увеличение представленности фрагментов, которые относятся к определенной хромосоме, теоретически должно свидетельствовать о трисомии по этой хромосоме. На практике все сложнее. Геном очень неоднороден и полон повторов, существуют участки, которые могут читаться чаще других, необходимо учитывать генетическое разнообразие человека и многое другое. Все это может приводить к ошибке во время подсчета равномерности распределения полученных на секвенаторе чтений. Поэтому при разработке алгоритмов анализа используются различные статистические модели, а входные данные тщательно фильтруются и корректируются.
Надо жить
Технология также предусматривает возможность определения инсерций и делеций отдельных участков хромосом при увеличении объема генерируемых секвенатором данных. Эти отклонения встречаются гораздо реже, чем трисомии, но не менее значимы для здоровья ребенка; при инвазивных обследованиях они могут остаться незамеченными. Существенно и то, что генетические пренатальные тесты изначально ориентированы на компьютерные расчеты, что позволяет исключить «человеческий фактор» как возможный источник ошибочных результатов.
Гораздо лучше, чем скрининг
В Федеральном научном центре акушерства гинекологии и перинатологии имени академика В.И. Кулакова в 2014 году при поддержке Министерства образования и науки и Министерства здравоохранения проведена валидация метода высокопроизводительного секвенирования (NGS) для неинвазивной пренатальной диагностики генетических нарушений у плода. Обследовано более 200 беременных с высоким риском анеуплоидий у плода на сроке 10-20 недель беременности. Метод продемонстрировал высокую чувствительность и специфичность (93% и 99% соответственно) при выявлении анеуплоидий по 21, 18 и X хромосомам (синдромы Дауна, Эдвардса, Тернера), что существенно превосходит по диагностическим характеристикам существующие неинвазивные скрининговые методы.
Основываясь на проведенном исследовании, можно рекомендовать использование высокопроизводительного секвенирования в качестве теста для неинвазивного скрининга на анеуплоидии 21, 18 и Х хромосом с последующим подтверждением положительных результатов инвазивными методами. Это позволит существенно сократить количество инвазивных диагностических процедур, сопряженных с риском потери беременности.—Денис Ребриков, доктор биологических наук, директор по науке «ДНК-Технология», научный сотрудник лаборатории молекулярно-генетических методов ФГУ НЦАГиП им. В.И.Кулакова
СИНДРОМ ЭДВАРДСА КАК ПРОЯВЛЕНИЕ ГЕНЕТИЧЕСКОГО ЗАБОЛЕВАНИЯ В ПЕДИАТРИЧЕСКОЙ ПРАКТИКЕ (клинический случай) Текст научной статьи по специальности «Клиническая медицина»
JOURNAL OF NEW MEDICAL TECHNOLOGIES, eEdition — 2018 — N 2
УДК:61 DOI: 10.24411/2075-4094-2018-16008
СИНДРОМ ЭДВАРДСА КАК ПРОЯВЛЕНИЕ ГЕНЕТИЧЕСКОГО ЗАБОЛЕВАНИЯ В ПЕДИАТРИЧЕСКОЙ ПРАКТИКЕ
(клинический случай)
Е.С. ЗАХАРОВА, А.Д. ЛАРИКОВА
ФГБОУ ВПО «Тульский государственный университет», медицинский институт, ул. Болдина, д. 128, Тула, 300028, Россия
Аннотация. В данной статье освещена тема редких генетических заболеваний в практике врача-педиатра на примере синдрома Эдвардса. Также представлен разбор клинического случая выявления данного синдрома у ребенка, находившегося на стационарном лечении в отделении выхаживания недоношенных детей. Врачу-педиатру необходимо и очень важно помнить о наличии такого грозного заболевания для своевременной диагностики и выбора тактики дальнейшего ведения таких пациентов. В статье представлены данные об этиологии, эпидемиологии, клинических признаках, диагностике и дальнейшей тактики лечения данной группы пациентов.
Синдром Эдвардса (другое название-синдром трисомии хромосомы 18) — генетическое заболевание, обусловленное наличием у человека дополнительной копии восемнадцатой хромосомы, то есть вместо двух 18-х хромосом в норме у больного присутствуют три 18-х хромосомы. Синдром Эдвардса является вторым по частоте хромосомным заболеванием после синдрома Дауна и характеризуется множеством пороков внутриутробного развития ребенка.
Описание основных симптомов данной болезни было сделано еще в начале XX века. До середины 1900-х годов собрать достаточную информацию об этой патологи не представлялось возможным. Во-первых, для этого необходим был соответствующий уровень развития технологий, который позволил бы обнаружить лишнюю хромосому. Во-вторых, большинство детей умирало в первые дни или недели жизни из-за низкого уровня оказания медицинской помощи. Первое полное описание болезни и ее основной причины (появление лишней 18-й хромосомы) было сделано только в 1960 году врачом Джоном Эдвардом, в честь которого тогда и назвали новую патологию.
Дети с синдромом Эдвардса имеют несовместимые с жизнью пороки развития внутренних органов, поэтому основная масса из них не доживает и до одного года. Более половины больных детей умирают в возрасте 2-3 месяцев, а до 12 месяцев доживает лишь около 10% пациентов. В настоящее время разработана государственная программа, включающая 24 стандарта оказания помощи больным с редкими заболеваниями, угрожающими жизни и приводящими к инвалидности.
Ключевые слова: синдром Эдвардса, дети, наблюдение, лечение.
THE EDWARDS SYNDROME AS THE MANIFESTATION OF GENETIC DISEASE IN PEDIATRIC
PRACTICE (clinical case)
E.S. ZAKHAROVA, A.D. LARICOVA
Tula State University, Medical Institute, Boldin Str., 128, Tula, 300028, Russia
Abstract. This article highlights the topic of rare genetic diseases in the practice of a pediatrician using the example of the Edwards syndrome. The authors present an analysis of the clinical case of the detection of this syndrome in a child who was hospitalized in the department for nursing preterm infants. The pediatrician should be aware of the presence of such a formidable disease for timely diagnosis and for choosing tactics for further management of such patients. The article presents data on etiology, epidemiology, clinical signs, diagnosis and further treatment tactics for this group of patients.
The Edwards syndrome (another name — the syndrome of trisomy chromosome 18) is a genetic disease caused by the presence of an additional copy of the eighteenth chromosome, that is, instead of two 18-chromosomes, the patient has three 18 chromosomes. The Edwards syndrome is the second most frequent chromosomal disease after the Down syndrome and it is characterized by a number of defects in the intrauterine development of the child.
The description of the main symptoms of this disease was made at the beginning of the XX century. Until the mid-1900s, it was not possible to gather sufficient information about this pathology. First, this requires an appropriate level of technology development, which would allow us to detect an extra chromosome. Secondly, most children died in the first days or weeks of life because of the low level of medical care. The first complete
JOURNAL OF NEW MEDICAL TECHNOLOGIES, eEdition — 2018 — N 2
description of this disease and its main cause (the appearance of an extra 18th chromosome) was made only in 1960 by a physician John Edward, in whose honor the new pathology was named.
Children with the Edwards syndrome have incompatible life-threatening malformations of internal organs, so most of them do not live up to one year. More than half of the sick children die at the age of 2-3 months, and up to 12 months only about 10% of patients survive. Currently, a state program has been developed, which includes 24 standards for the care of patients with rare diseases that threaten life and lead to disability.
Key words: the Edwards syndrome, children, observation, treatment.
Синдром Эдвардса — хромосомное заболевание, обусловленное трисомией по 18-ой хромосоме и сопровождающееся множественными пороками развития. Частота встречаемости синдрома Эдвардса варьирует от 1 на 3000-8000 рождённых [2-4].
В патогенезе развития синдрома Эдвардса выступает наличие дополнительной 18-й хромосомы в кариотипе зиготы. Лишняя хромосома у гамет появляется, как правило, из-за нерасхождения хромосом в случае мейотического деления, в результате чего в половой клетке оказывается 24 хромосомы. Если при оплодотворении такой клеткой встречается гамета от противоположного пола, то ими образуется зигота с трисомией.
Существуют следующие вариации: мозаичная трисомия (наличие лишней хромосомы не во всех клетках организма), частичная трисомия (когда присутствует только часть лишней хромосомы), полная трисомия (когда ребенок наследует полную дополнительную копию лишней хромосомы) [4, 6, 7].
Для синдрома Эдвардса характерны: своеобразные фенотипические признаки, аномалии опорно-двигательной системы, аномалии сердечно-сосудистой системы, аномалии пищеварительной системы, аномалии мочеполовой системы, пороки развития ЦНС.
Фенотипические признаки: долихоцефалическая форма черепа, низкий лоб, выступающий затылок, микрогнатия, маленький рот, микрофтальмия, расщелины верхней губы и нёба, эпикантус, птоз, экзофтальм, косоглазие, короткая шея с избыточной кожной складкой, деформации ушных раковин.
Пороки развития опорно-двигательной системы: скрещенные пальцы кистей, укороченная грудина, аномалии ребер, врожденный вывих бедра, косолапость, «стопа-качалка», синдактилия стоп.
Пороки развития сердечно-сосудистой системы: дефекты межжелудочковой и межпредсердной перегородок, коарктация аорты, транспозиция магистральных сосудов, дисплазия клапанов, тетрада Фалло, аномальный дренаж легочных вен, декстракардия.
Пороки развития пищеварительной системы: диафрагмальные, пупочные и паховые грыжи, дивертикул Меккеля, трахеопищеводные свищи, пилоростеноз, атрезия подвздошной кишки и ануса.
Пороки развития мочеполовой системы: подковообразная почка, гидронефроз, дивертикулы мочевого пузыря, гипоспадия и крипторхизм (у мальчиков), двурогая матка, внутриматочная перегородка и гипертрофия клитора (у девочек).
Пороки развития ЦНС: микроцефалия, менингомиелоцеле, гидроцефалия, аномалии Арнольда-Киари, кисты арахноидального сплетения, гипоплазия мозжечка и мозолистого тела [1, 2, 4, 5].
Диагностика включает в себя: УЗИ плода и доплерография маточно-плацентарного кровотока (в ранних сроках беременности можно заподозрить пороки развития головного мозга и конечностей, также наличие обильного количества околоплодной жидкости), анализ крови на сывороточные маркеры (в-субъединицы хорионического гонадотропина (РХГ), а-фетопротеина (АФП), эстриола (ЕЗ), 17-окси прогестерона), инвазивная дородовая диагностика (биопсия хориона, амниоцентез, кордоцентез) с последующим кариотипированием плода.
Поскольку в большинстве случаев аномалии развития оказываются несовместимыми с жизнью, лечение детей с синдромом Эдвардса сводится к оказанию симптоматической помощи, направленной на поддержание физиологических функций, продление жизни и улучшение ее качества. Хирургическая коррекция врожденных пороков, как правило, является рискованной и неоправданной [2, 5, 7].
Поскольку дети с синдромом Эдвардса ослаблены и подвержены частой заболеваемости инфекциями мочевыводящих путей, средним отитом, конъюнктивитом, синуситами, пневмонией, они нуждаются в тщательно организованном уходе, полноценном питании, регулярном наблюдении педиатра [1, 2, 8].
Представляем клинический случай синдрома Эдвардса у ребенка в возрасте 1 мес. 25 суток.
Под наблюдением в течение 14 дней находился ребенок в возрасте 1 мес. 25 суток. Ребенок переведен из ОРИТ в отделение выхаживания недоношенных детей с диагнозом: Множественные врожденные пороки развития: агенезия мозолистого тела. Единственная почка слева. Перинатальное гипокси-чески — ишемическое поражение ЦНС, с-м угнетения. Пневмопатия, ст. разрешения, ДН 0-1 ст. Неона-тальная желтуха. Открытое овальное окно, НК 0 ст. Дисплазия тазобедренных суставов. ЗВУР по гипоксическому типу. Внутриутробное инфицирование.
Из анамнеза жизни известно: роды 2 срочные в 39 недель, экстренно-оперативным путем кесарева сечения. Поперечное положение плода. Хр. плацентарная недостаточность плода. Анемия беременных. Хр. АГ. Ожирение I ст. При рождении оценка по шкале Апгар на 1 минуте 5 б., на 5 минуте 6
JOURNAL OF NEW MEDICAL TECHNOLOGIES, eEdition — 2018 — N 2
баллов, вес — 1590 г, рост — 44 см. Окружность головы 31 см, груди — 30 см. Первичная помощь в род. зале — санация ВДП, рИВЛ через маску 3 минуты + увл. кислородом. Состояние при рождении: тяжелое, обусловленное ДН 1 степени, неврологическим статусом, степенью внутриутробной гипотрофии, внутриутробным инфицированием. По тяжести состояния здоровья ребенок переведен в ОРИТ. Проводимая терапия в ОРИТ: дыхательная поддержка, антибиотикотерапия, инфузионная терапия, фототерапия 2-7 суток, режим кувезный. На фоне проводимого лечения состояние стабилизировалось. Ребенок переведен в отделение выхаживания недоношенных детей с диагнозом:
Множественные врожденные пороки развития: агенезия мозолистого тела. Единственная почка слева. Перинатальное гипоксически — ишемическое поражение ЦНС, с-м угнетения. Пневмопатия, ст. разрешения, ДН 0-1 ст. Неонатальная желтуха. Открытое овальное окно, НК 0 ст. Дисплазия тазобедренных суставов. ЗВУР по гипоксическому типу. Внутриутробное инфицирование.
Проведено обследование:
Общий анамнез мочи в динамике: от 7.10: норма. От 15.10, 25.10: дрожжевые грибы
Кал на УПФ 15.10: патогенная флора не выявлена.
Анализ крови на кариотип от 10.10: Синдром Эдвардса, трисомная форма.
Rg грудной клетки в динамике: от 7.10: легочные поля прозрачные, легочный рисунок усилен в прикорневых зонах. Средостение не смещено.
Rg грудной клетки от 22.10: легочные поля прозрачные, легочный рисунок б/о.
НСГ в динамике: от 7.10: значительные признаки гипоксии, незрелости. Аномалия строения головного мозга: агенезия мозолистого тела. Дилатация тел и задних рогов боковых желудочков I-II ст. Псевдокисты: ПВК I-II ст. слева и сосудистого сплетения справа. Дилатация цистерны мозга. НСГ от 21.10: без динамики.
ЭХО-КГ от 7.10: ООО 3 мм. Аномалия строения АоК
УЗИ брюшной полости от 10.10: единственная галетообразная почка слева. Деформация желчного пузыря.
УЗИ тазобедренных суставов от 13.10: умеренная степень дисплазии.
ЭКГ от 8.10: ритм синусовый, правильный. ЭОС — полувертикальное положение.
Окулист от 13.10: преретинопатия (высокий риск прогрессирования).
Невролог от 14.10: гипоксически-ишемическое поражение ЦНС
Ортопед от 25.10: дисплазия тазобедренных суставов.
Генетик от 10.10: Синдром Эдвардса, трисомная форма.
Лечение, проводимое в отделении выхаживания недоношенных детей.
— Антибактериальная терапия (Цефомакс 80 мг в/в 2 раза в день)
— Профилактика и лечение капиллярных кровотечений (Этамзилат 2,5% 0,3 мл в/в 2 раза в день)
— Улучшение метаболизма (Элькар 30% 5 кап. per os 2 раза в день)
— Профилактика дисбактериоза (Бифидумбактерин 2,5 дозы per os 2 раза в день)
— Витамины А (1 кап. 1 раз в день per os) и Е(1 кап. 1 раз в день per os)
— Эмоксипин 1 % (1 кап. 6 раз/час) 1 раз в сутки в глаза.
После отделения выхаживания недоношенных детей при достижении массы тела 2500 гр. ребенок был переведен в областной дом ребенка.
Большинство детей, которые родились с синдромом Эдвардса, не доживают до первого года своей жизни. Средняя продолжительность жизни для половины детей, рожденных с этим синдромом, менее чем два месяца. От девяноста до девяноста пяти процентов из этих детей умирает, не дожив до своего первого дня рождения. От пяти до десяти процентов детей, которые выжили в первый год, испытывают серьезные отклонениями в развитии.
Разработана государственная программа по профилактике орфанных заболеваний (закон РФ от 21 ноября 2011 г. № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации»), которая содержит следующие пункты: планирование беременности, контроль за состоянием здоровья женщины, витаминотерапия в течение беременности, регулярное наблюдение беременной в условиях женской консультации.
Литература
1. Вахарловский В.Г. Генетика в практике педиатра. Руководство для врачей. Изд-во СПбГПМА, 2009. 286 с.
2. Вахарловский В.Г., Горбунова В.Н. Клиническая генетика. СПб.: Изд-во СПбГПМА, 2010. №38.
3. Иванов В.И. Генетика: учебник для вузов / под ред. акад. Иванова В.И. М.: ИКЦ «Академкнига», 2009. 638 с.
4. Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. М., 2010. 448 с.
ВЕСТНИК НОВЫХ МЕДИЦИНСКИХ ТЕХНОЛОГИЙ, электронный журнал — 2018 — N 2 JOURNAL OF NEW MEDICAL TECHNOLOGIES, eEdition — 2018 — N 2
5. Курзина E.A. Роль наследственных заболеваний у детей с перинатальной патологией // Вестник Педиатрической академии. 2009. № 5. С. 96-101.
6. Новиков П.В. Семиотика наследственных болезней у детей (симптом-синдром-болезнь). М.: Триада-Х, 2009. 432 с .
7. Таточенко В.К. Педиатру на каждый день. Справочник по диагностике и лечению. Издание восьмое, дополненное. М., 2016. 271 с.
8. Шабалов Н.П. Справочник педиатра. 3-е издание П., 2015. 577 с.
References
1. Vakharlovskiy VG. Genetika v praktike pediatra [Genetics in the practice of pediatrician. A guide for physicians]. Rukovodstvo dlya vrachey. Izd-vo SPbGPMA; 2009. Russian.
2. Vakharlovskiy VG, Gorbunova VN. Klinicheskaya genetika [Clinical genetics]. Sankt-Peterburg: Izd-vo SPbGPMA; 2010. Russian.
3. Ivanov VI. Genetika: uchebnik dlya vuzov [Genetics: textbook for universities]. Pod red. akad. Ivanova VI. Moscow: IKTs «Akademkniga»; 2009. Russian.
4. Kozlova SI, Demikova NS. Nasledstvennye sindromy i mediko-geneticheskoe konsul’tirovanie [Hereditary syndromes and medical genetic counseling]. Moscow; 2010. Russian.
5. Kurzina EA. Rol’ nasledstvennykh zabolevaniy u detey s perinatal’noy patologiey [the role of hereditary diseases in children with perinatal pathology]. Vestnik Pediatricheskoy akademii. 2009;5:96-101. Russian.
6. Novikov PV. Semiotika nasledstvennykh bolezney u detey (simptom-sindrom-bolezn’) [Semiotics of hereditary diseases in children (symptom-syndrome-disease)]. Moscow: Triada-Kh; 2009. Russian.
7. Tatochenko VK. Pediatru na kazhdyy den’. Spravochnik po diagnostike i lecheniyu. [the Pediatrician every day. Guide to diagnosis and treatment] Izdanie vos’moe, dopolnennoe. Moscow; 2016. Russian.
8. Shabalov NP. Spravochnik pediatra [Handbook of pediatrician]. 3-e izdanie P.; 2015. Russian.
Библиографическая ссылка:
Захарова Е.С., Ларикова А.Д.Синдром Эдвардса как проявление генетического заболевания в педиатрической практике (клинический случай) // Вестник новых медицинских технологий. Электронное издание. 2018. №2. Публикация 1-3. URL: http://www.medtsu.tula.ru/VNMT/Bulletin/E2018-2/1-3.pdf (дата обращения: 15.03.2018). DOI: 10.24411/2075-4094-2018-16008.
Синдром Эдвардса (трисомия 18) — NHS
Синдром Эдвардса, также известный как трисомия 18, является редким, но серьезным заболеванием.
Синдром Эдвардса влияет на продолжительность жизни ребенка. К сожалению, большинство детей с синдромом Эдвардса умирают до или вскоре после рождения.
Небольшое количество (около 13 из 100) детей, рожденных живыми с синдромом Эдвардса, доживут до своего первого дня рождения.
Причина синдрома Эдвардса
Каждая клетка вашего тела обычно содержит 23 пары хромосом, которые несут гены, унаследованные вами от родителей.
У ребенка с синдромом Эдвардса 3 копии хромосомы номер 18 вместо 2. Это влияет на то, как ребенок растет и развивается. Наличие 3 копий 18 хромосомы обычно происходит случайно из-за изменения сперматозоидов или яйцеклетки до зачатия ребенка.
Ваш шанс родить ребенка с синдромом Эдвардса увеличивается с возрастом, но любой может иметь ребенка с синдромом Эдвардса. Состояние обычно не передается в семье и не вызвано чем-либо, что родители сделали или не сделали.
Обратитесь к терапевту, если хотите узнать больше. Они могут направить вас к генетическому консультанту.
Типы синдрома ЭдвардсаСимптомы и степень серьезности поражения вашего ребенка обычно зависят от того, есть ли у него полный, мозаичный или частичный синдром Эдвардса.
Полный синдром Эдвардса
Большинство детей с синдромом Эдвардса имеют дополнительную 18-ю хромосому, присутствующую во всех клетках. Это называется полным синдромом Эдвардса.
Последствия полного синдрома Эдварда часто бывают более серьезными.К сожалению, большинство детей с этой формой умирают до того, как родятся.
Мозаика Синдром Эдвардса
Небольшое количество детей с синдромом Эдвардса (примерно 1 из 20) имеют лишнюю 18-ю хромосому лишь в некоторых клетках. Это называется мозаичным синдромом Эдвардса (или иногда мозаичной трисомией 18).
Это может привести к более мягким последствиям заболевания, в зависимости от количества и типа клеток, у которых есть дополнительная хромосома. Большинство рожденных живыми младенцев с этим типом синдрома Эдварда проживут не менее года и могут дожить до 9000 лет.
Частичный синдром Эдвардса
У очень небольшого числа детей с синдромом Эдвардса (примерно 1 из 100) в клетках имеется только часть дополнительной хромосомы 18, а не целая дополнительная хромосома 18. Это называется частичной хромосомой. Синдром Эдвардса (или иногда частичная трисомия 18).
Как частичный синдром Эдвардса влияет на ребенка, зависит от того, какая часть хромосомы 18 присутствует в его клетках.
Консультации для молодых родителей
Есть поддержка для всего, что нужно вам или вашему ребенку.
Все дети, рожденные с синдромом Эдвардса, будут иметь некоторую неспособность к обучению.
Синдром Эдвардса связан с определенными физическими особенностями и проблемами со здоровьем. Каждый ребенок уникален и будет иметь разные проблемы со здоровьем и потребности. Обычно они имеют низкий вес при рождении, а также могут иметь широкий спектр физических симптомов. У них также могут быть сердечные, респираторные, почечные или желудочно-кишечные заболевания.
Несмотря на свои сложные потребности, дети с синдромом Эдвардса могут постепенно начать делать больше вещей.
Как и любой ребенок, они:
- обладают собственной индивидуальностью
- учатся в своем собственном темпе
- обладают важными и уникальными для них вещами
Старайтесь не думать слишком далеко и проводить время с ребенком .
Скрининг синдрома Эдвардса
Если вы беременны, вам предложат пройти обследование на синдром Эдвардса на сроке от 10 до 14 недель беременности. Это проверяет вероятность того, что у вашего ребенка это заболевание.
Этот скрининговый тест называется комбинированным тестом, и он определяет вероятность наличия у ребенка синдрома Эдвардса, синдрома Дауна и синдрома Патау.
Во время теста вам сделают анализ крови и ультразвуковое сканирование, чтобы измерить жидкость в задней части шеи вашего ребенка (затылочная прозрачность).
Подробнее о скрининге на синдром Эдвардса на 10–14 неделе
Если невозможно измерить жидкость в задней части шеи вашего ребенка, или если вы беременны на сроке более 14 недель, вам предложат скрининг на Синдром Эдвардса как часть вашего 20-недельного сканирования.Это иногда называют сканированием в середине беременности. Это ультразвуковое сканирование, позволяющее определить, как растет ваш ребенок.
Скрининг не может определить, какая форма синдрома Эдвардса может быть у вашего ребенка или как это повлияет на него.
Подробнее о 20-недельном сканировании
Диагностика синдрома Эдвардса во время беременности
Если комбинированный тест показывает, что у вас больше шансов родить ребенка с синдромом Эдвардса, вам будет предложено пройти тест, чтобы точно определить, есть ли у вашего ребенка это заболевание.
Этот диагностический тест включает в себя анализ образца клеток вашего ребенка, чтобы проверить, есть ли у него дополнительная копия хромосомы 18.
Есть 2 разных способа получить этот образец клеток:
Это инвазивные тесты, которые увеличивают ваши шансы выкидыш. Ваш врач обсудит это с вами.
Результаты диагностического теста
Врач-специалист (акушер) или акушерка объяснит, что означают результаты скрининга, и расскажет вам о возможных вариантах.
Это очень сложная ситуация, и нормально испытывать целый ряд эмоций. Может быть полезно поговорить со своим врачом, семьей и друзьями или партнером о том, о чем вы думаете и что чувствуете.
Если вам сказали, что у вашего ребенка синдром Эдвардса, до рождения или после него, вам будет предложена поддержка и информация.
Вы можете посетить веб-сайт SOFT UK, чтобы получить поддержку и дополнительную информацию о синдроме Эдвардса, а также связаться с другими семьями, затронутыми этим заболеванием.
Вы также можете связаться с Antenatal Results and Choices (ARC), где есть информация о скрининговых тестах и о том, что вы можете почувствовать, если вам скажут, что у вашего ребенка есть или могут быть проблемы.
У ARC есть телефон доверия, на который можно позвонить по телефону 0845077 2290 или 0207713 7486 с мобильного телефона с понедельника по пятницу с 10:00 до 17:30. На горячую линию отвечает обученный персонал, который может предложить информацию и поддержку.
Подробнее о том, что происходит, если антенатальные скрининговые тесты что-то обнаруживают
Диагностика синдрома Эдвардса после рождения
Если врачи считают, что у вашего ребенка синдром Эдвардса после рождения, будет взят образец крови, чтобы проверить, есть ли дополнительные копии хромосомы 18.
Лечение синдрома Эдвардса
От синдрома Эдвардса нет лекарства.
Лечение будет сосредоточено на симптомах заболевания, таких как сердечные заболевания, затрудненное дыхание и инфекции. Вашему ребенку также может потребоваться кормление через зонд, так как у него часто возникают трудности с кормлением.
Синдром Эдвардса влияет на движения вашего ребенка, когда он становится старше, и ему может быть полезно поддерживающее лечение, такое как физиотерапия и трудотерапия.
В зависимости от конкретных симптомов вашего ребенка, ему может потребоваться помощь специалиста в больнице или хосписе, или вы сможете ухаживать за ним дома с необходимой поддержкой.
Советы опекунам
Поддержка человека с синдромом Эдвардса может быть одновременно полезным и трудным.
Если вам нужна помощь или вы просто хотите поговорить с кем-нибудь, есть много возможностей поддержки.
В вашем справочнике по социальной помощи и поддержке содержится множество советов о том, как вы можете найти время, чтобы позаботиться о себе, в том числе:
- перерыв в уходе
- получение юридической поддержки и защиты
- забота о своем благополучии
Поговорите с родителями и семьями
Это может помочь поговорить с другими родителями и семьями, которые знают, как вы себя чувствуете.
Вы можете сделать это:
- связаться с людьми на форумах и в социальных сетях
- пойти в группу поддержки — спросите свою акушерку или патронажную сестру о доступных группах поддержки
Информация о вашем ребенке
Если у вашего ребенка будет обнаружен синдром Эдвардса до или после его рождения, его клиническая бригада передаст информацию о нем в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать более эффективные способы лечения симптомов заболевания. Вы можете отказаться от регистрации в любое время.
Узнайте больше о реестре на GOV.UK
Последняя проверка страницы: 25 сентября 2020 г.
Срок следующей проверки: 25 сентября 2023 г.
Что такое трисомия 18? — Trisomy 18 Foundation
Трисомия 18, также известная как синдром Эдвардса, — это состояние, вызванное ошибкой в делении клеток, известной как мейотическая дизъюнкция.Когда это происходит, вместо нормальной пары появляется дополнительная хромосома 18 (тройная) в развивающемся ребенке и существенно нарушает нормальный паттерн развития, что может быть опасно для жизни, даже до рождения. Ошибка трисомии 18 встречается примерно в 1 из каждых 2500 беременностей в США и в 1 из 6000 живорождений. Общее количество родов намного выше, потому что оно включает значительное количество мертворождений, которые происходят во 2-м и 3-м триместрах беременности.
В отличие от синдрома Дауна, который также вызывается лишней хромосомой, проблемы развития, вызванные трисомией 18, связаны с большим количеством медицинских осложнений, которые потенциально опасны для жизни в первые месяцы и годы жизни.Исследования показали, что только 50% доношенных младенцев будут рождены живыми, а у девочек будет более высокий процент живорождений, чем у мальчиков.
При рождении поступление в отделения интенсивной терапии в отделения интенсивной терапии новорожденных (ОИТН) является обычным делом для младенцев с трисомией 18. Опять же, мальчики будут иметь более высокий уровень смертности в этот неонатальный период, чем девочки, хотя дети с более высокой массой тела при рождении чувствуют себя лучше во всех случаях категории.
Некоторые младенцы смогут выжить и будут выписаны из больницы с помощью ухода на дому для оказания помощи родителям.И хотя 10 или более процентов могут дожить до своих первых дней рождения, есть дети с трисомией 18, которые могут прожить много лет со своими семьями, достигая определенных результатов и участвуя в жизни своего сообщества. Небольшое количество взрослых (обычно девочек) с трисомией 18 имеют и доживают до двадцати и тридцати лет, хотя и со значительными задержками в развитии, которые не позволяют им жить самостоятельно без постоянного ухода.
Что такое связанные условия? — Trisomy 18 Foundation
Самая распространенная трисомия — это трисомия 21, также известная как синдром Дауна, когда у ребенка три хромосомы из двадцать первой.Трисомия 18 является второй по распространенности трисомией и возникает, когда у ребенка три из восемнадцатой хромосомы. Это приводит к 47 хромосомам вместо нормальных 46 в пораженных клетках. Именно этот дополнительный генетический материал вызывает проблемы, связанные с трисомией 18. Третьей по распространенности является трисомия 13, также известная как синдром Патау.
Хотя существуют разные типы трисомии 18, это не означает, что один для ребенка лучше, чем другой. У каждого типа есть ряд возможностей.Некоторые дети слабы с медицинской точки зрения, в то время как другие процветают; некоторые дети ходят пешком, а другие прикованы к инвалидным коляскам. Трудно сказать, как дополнительная хромосома повлияет на конкретного ребенка, исходя только из одного генетического диагноза.
Типы трисомии 18:
- Полная трисомия 18: Наиболее распространенным типом трисомии 18 (встречающейся примерно в 95% всех случаев) является полная трисомия. При полной трисомии дополнительная хромосома встречается в каждой клетке тела ребенка. Этот тип трисомии не передается по наследству.Это не из-за того, что родители сделали или не сделали — до или во время беременности.
- Частичная трисомия 18: Частичная трисомия очень редка. Они возникают, когда присутствует только часть дополнительной хромосомы. Некоторые синдромы частичной трисомии 18 могут быть вызваны наследственными факторами. Очень редко кусок 18 хромосомы прикрепляется к другой хромосоме до или после зачатия. У больных есть две копии 18-й хромосомы плюс «частичный» кусок дополнительного материала 18-й хромосомы.
- Мозаичная трисомия 18: Мозаичная трисомия также встречается очень редко. Это происходит, когда дополнительная хромосома присутствует в некоторых (но не во всех) клетках тела. Как и полная трисомия 18, мозаичная трисомия не передается по наследству и возникает случайно во время деления клеток.
Трисомия 18 — NORD (Национальная организация по редким заболеваниям)
Jorde LB, Carey JC и Bamshad MJ. Электронная книга по медицинской генетике. 2015. Elsevier Health Sciences.
Кэссиди С.Б. и Аллансон Дж.Управление генетическими синдромами. 2010. Джон Вили и сыновья.
Кэри Дж. Синдром трисомии 18. В: Справочник НОРД по редким заболеваниям. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003: 88.
СТАТЬИ В ЖУРНАЛЕ
Farmakis SG, Barnes AM, et al. Рекомендации по скринингу солидных опухолей при трисомии 18. Am J Med Genet. 2019; 179A: 455-466.
Жанвье А., Фарлоу Б., Баррингтон К.Дж., Бурк С.Дж., Бразг Т. и Уилфонд Б. Укрепление доверия и улучшение общения с родителями детей с трисомией 13 и 18: исследование с использованием смешанных методов.Паллиативная медицина 2019; 0269216319860662.
Като Е., Китасе Ю., Татибана Т. и др. Факторы, связанные с выживанием при трисомии 18: ретроспективное многоцентровое исследование. Американский журнал медицинской генетики, часть A. 2019; 179: 1253–1259.
Тайра Р., Иноуэ Х, Савано Т. и др. Ведение апноэ у младенцев с трисомией 18. Медицина развития и детская неврология 2019: 25 ноября. Doi: 10.1111 / dmcn.14403. [Epub перед печатью]
Алькхамди М.А., Диого Р. и др. Подробное исследование опорно-двигательного аппарата плода с трисомией-18 (синдром Эдвардса) с подмышечной дугой Лангера и сравнение с другими случаями врожденных пороков развития человека.J Anat Sci Res. 2018; 1: 1-8.
Инуое А., Сузуки Р. и др. Терапевтический опыт лечения гепатобластомы, связанной с трисомией 18. Рак крови у детей. 2018; 65: e27093.
Петерсон Р., Каламур Н., Фиоре А., Хаддлстон С. и Спенс К. Факторы, влияющие на исходы после кардиологического вмешательства у младенцев с трисомией 13 и 18. Детская кардиология 2018; 39 (1): 140-147.
Kosiv KA, Gossett JM, Bai S, & Collins RT. Врожденные операции на сердце по поводу госпитальной смертности при трисомии 13 и 18.Педиатрия 2017; 140 (5): e20170772.
Паттерсон Ф. Тейлор Дж. И др. Переходы к лечению младенцев с трисомией 13 или 18. Amer J Perinatol. 2017; 34: 887-894.
Петерсон Дж. К., Кочилас Л. К., Каттон К. Г., Моллер Дж. Х. и Сетти С. П.. Отдаленные исходы детей с трисомией 13 и 18 после вмешательств по поводу врожденных пороков сердца. Анналы торакальной хирургии 2017; 103 (6): 1941-1949.
Andrews SE, Downey AG, et al. Совместное принятие решений и подходы в пренатальном и постнатальном ведении синдромов трисомии 13 и трисомии 18.Am J Med Genet C Semin Med Genet. 2016; 172: 257-263.
Брунс Д.А. и Мартинес А. Анализ пороков сердца и хирургических вмешательств в 84 случаях с полной трисомией 18. Американский журнал медицинской генетики, часть A, 2016 г .; 170 (2): 337-343.
Кэри Дж. С. и Барнс А. М.. Опухоль Вильмса и трисомия 18: есть ли связь? В Американском журнале медицинской генетики, часть C: Семинары по медицинской генетике, 2016 г .; 172 (3): 307-308.
Донован Дж. Х., Кригбаум Дж. И Брунс Д. А. Медицинские вмешательства и выживаемость детей с трисомией 18 по полу.В American Journal of Medical Genetics Part C, Seminars in Medical Genetics 2016; 172 (3): 272-278.
Josephsen JB, Armbrecht ES, Braddock SR, & Cibulskis CC. Процедуры на 1-м году жизни для детей с трисомией 13 и трисомией 18, 25-летний одноцентровый обзор. В American Journal of Medical Genetics Part C: Seminars in Medical Genetics 2016 ;. 172 (3): 264-271.
Meyer RE, Liu G, Gilboa SM и др. И Национальная сеть по предотвращению врожденных дефектов. Выживаемость детей с трисомией 13 и трисомией 18: популяционное исследование с участием нескольких штатов.Американский журнал медицинской генетики Part A 2016; 170 (4): 825-837.
Нельсон К.Е., Розелла Л.К. и др. Выживаемость и хирургические вмешательства у детей с трисомией 13 и 18. JAMA. 2016; 316: 420-428.
Сатге Д., Ниши М., Сирвент Н. и Векеманс М. Профиль опухоли при синдроме Эдвардса (трисомия 18). В American Journal of Medical Genetics Part C: Seminars in Medical Genetics 2016; 172 (3): 296-306.
Брунс Д.А. Статус развития 22 детей с трисомией 18 и восьми детей с трисомией 13: значение и рекомендации.Американский журнал медицинской генетики, часть A, 2015 г .; 167 (8): 1807-1815.
Cereda A & Carey JC. Синдром трисомии 18. Журнал редких заболеваний Orphanet 2012; 7 (1): 81.
Palomaki GE, Deciu C, et al. Секвенирование ДНК материнской плазмы надежно идентифицирует трисомию 18 и трисомию 13, а также синдром Дауна: международное совместное исследование. Genet Med. 2012; 14: 296–305.
ИНТЕРНЕТ
Трисомия 18. Информационный центр по генетическим и редким заболеваниям (GARD). Трисомия 18. Последнее обновление: 07.07.2015.https://rarediseases.info.nih.gov/diseases/6321/trisomy-18 По состоянию на 22 января 2020 г.
Синдром Эдвардса — симптомы, лечение и причины
На этой странице
Что такое синдром Эдвардса?
Синдром Эдвардса — это генетическое заболевание младенцев, вызывающее тяжелую инвалидность. Это вызвано дополнительной копией 18 хромосомы, и дети, рожденные с этим заболеванием, обычно не живут дольше недели.
Как возникает синдром Эдвардса?
У детей с синдромом Эдвардса есть 3 копии части или всей хромосомы 18 вместо обычных 2 копий.Ее еще называют Трисомия 18.
Это может быть вызвано ошибкой в формировании яйцеклетки или сперматозоидов, либо проблема может возникнуть во время развития ребенка в утробе матери.
Младенцы с синдромом Эдвардса либо выкидыши, либо мертворождены, либо рождаются с серьезными физическими отклонениями. Очень редко ребенок с синдромом Эдвардса доживает до первого года жизни, и большинство из них умирают в течение недели после рождения.
Синдром Эдвардса гораздо чаще встречается у девочек.
Каковы симптомы синдрома Эдвардса?
Младенцы с синдромом Эдвардса могут иметь:
- Низкая масса тела при рождении
- маленькая головка и челюсть
- необычное лицо и голова
- необычные кисти и ступни с перекрывающимися пальцами и перепонками на пальцах ног
- Проблемы с кормлением, дыханием, зрением и слухом
Как диагностируется синдром Эдвардса?
Синдром Эдвардса можно заподозрить или даже диагностировать во время беременности.
Иногда вероятность синдрома Эдвардса повышается после 11–13-недельных тестов (обычно УЗИ на беременность и анализ крови). Также возможно выявить синдром Эдвардса с помощью неинвазивного пренатального тестирования (НИПТ), которое представляет собой анализ крови, взятый на 10 неделе беременности.
Но единственный надежный способ диагностировать синдром Эдвардса — это генетическое тестирование. Это можно сделать, пока ребенок находится в утробе матери, с помощью биопсии ворсин хориона (CVS) или амниоцентеза.Оба этих теста имеют риск выкидыша. Вы можете поговорить об этом со своим акушером или акушеркой.
Иногда синдром Эдвардса не подозревается во время беременности и обнаруживается только при рождении ребенка.
Посетите веб-сайт по беременности, родам и младенцам для получения дополнительной информации о пренатальном скрининге.
Как лечится синдром Эдвардса?
Не существует эффективного длительного лечения детей с синдромом Эдвардса. Родители в этой ситуации сталкиваются с очень тяжелым выбором.Они могут оставить своих младенцев живыми в отделении интенсивной терапии новорожденных. Или они могут выбрать, чтобы их ребенок получал паллиативную помощь до тех пор, пока он или она не умрет.
Ресурсы и поддержка
Посетите наш справочник по генетическим заболеваниям, чтобы узнать больше о генах, типах генетических нарушений, а также о том, куда обратиться за помощью и получить дополнительную информацию.
Очень особенный день рождения для молодого человека с трисомией 18
Дополнительная 18-я хромосома разрушительна, но некоторые дети преодолевают трудности и выживают в младенчестве.
10 сентября Донни Хитон отметит свой 21-й день рождения. Но в отличие от большинства 21-летних, Донни весит всего 55 фунтов. Он — один из старейших известных людей с трисомией 18 (синдром Эдварда). Каждая из его клеток имеет дополнительную хромосому 18. Каждый из 6000 новорожденных страдает этим заболеванием.
Трисомия 18 была в новостях во время последних президентских выборов, потому что она есть у маленькой дочери Рика Санторума Изабеллы. Ее маме было 48 лет, когда она родила Изабеллу в 2008 году; Маме Донни было 42 года, когда он родился.Риск рождения яйца с лишней хромосомой увеличивается с возрастом матери.
Я использую довольно ужасный случай, чтобы проиллюстрировать трисомию 18 в моем учебнике по генетике человека, мальчик, который умер в возрасте 22 дней. А Google images вытаскивает фотографии плодов, которые так и не появились.
Я только несколько раз сталкивался с трисомией 18 при генетическом консультировании, когда дополнительная хромосома обнаруживалась при пренатальном тестировании у женщин «пожилого материнского возраста» — старше 35 лет. Эти случаи заканчивались прерыванием беременности естественным путем или по собственному желанию.Мы вызвали священника по одному случаю, который откровенно сказал, что прервет беременность, настолько мрачный прогноз. В другом случае пара потеряла ребенка в 4 месяца, и этот эффект был разрушительным для их тогдашнего дошкольника, который потерял своего младшего брата. Беременные в третий раз, пара хотела провести амниоцентез, чтобы не иметь еще одного ребенка с этим заболеванием, хотя эмпирический риск рецидива составляет только 1 из 100.
Трисомия не передается по наследству; дополнительная хромосома — это случайность мейоза, пара хромосом, которая не разделяется при формировании гамет, а вместо этого перегружает оплодотворенную яйцеклетку.Технически это называется не дизъюнкцией, и это просто случается — никто не виноват.
ПРЕНАТАЛЬНОЕ ТЕСТИРОВАНИЕ
Сегодня, благодаря внеклеточному тестированию ДНК плода (также известному как неинвазивная пренатальная диагностика), многие пациенты могут избежать небольшого риска амниоцентеза и взятия проб ворсинок хориона и узнать о наиболее распространенных аутосомных трисомиях (13, 18, и 21). ДНК плода перемещается в кровотоке матери в частях меньшего размера, чем ДНК будущей мамы, поэтому ее можно разделить и сравнить.Если количество частей хромосомы 18 превышает количество частей других хромосом вдвое, трисомия 18 уже под рукой.
Первые две версии этого типа тестов — Harmony и Materni-T, стали доступны в 2011 году и вызвали шквал заголовков. В конце 2012 года Американский колледж акушеров и гинекологов высоко оценил эти тесты, но пока только для женщин из группы высокого риска, которые в противном случае проходили бы более инвазивные тесты. Но дальнейшие исследования могут вскоре прояснить это для всех. Незадолго до этого в блоге DNA Science объяснялось, как в кровотоке беременных женщин содержится ДНК плода.
Пренатальное тестирование стало настолько распространенным, что, возможно, мы упускаем из виду тот факт, что выявляемые медицинские состояния могут быть разными. Не все плоды, новорожденные и малыши проигрывают битву с трисомией 18. Не все решают избежать рождения ребенка с трисомией 18, и не все выбирают пренатальный диагноз. Карен Хитон, мама Донни, отказалась от пренатального тестирования. А ее сыну, который, по прогнозам врачей, не проживет больше нескольких месяцев, скоро исполнится 21 год. Я прочитал о Донни в потрясающей статье в Tampa Bay Times штатного писателя Лейна ДеГрегори, а затем поговорил с Карен.
При трисомии 18 пальцы явно перекрываются.21-летний Донни — исключение. Половина плодов с трисомией 18 не доживает до рождения, и менее 5 процентов детей достигают первых дней рождения, большинство из них имеют серьезные проблемы: дыры в сердце, дефекты почек, пищевод, не связанный с желудком. Рост задерживается, и ребенок никогда не продвигается дальше шести месяцев в своем развитии. Голова небольшая, с крохотной челюстью и низко посаженными ушами. Два характерных признака — это «ступни качалки» и странное наложение пальцев в крошечных сжатых кулаках.
Но некоторые дети с синдромом выросли в юном возрасте. Мать Донни Карен посвятила ему свою жизнь. Вот часть ее истории.
ВЗГЛЯД МАТЕРИ
«Мне было 42 года, когда я забеременела Донни. У меня не было амниоцентеза, потому что я не верю в аборты и не думала, что что-то будет неправильно. Я думал, что он будет здоровым подпрыгивающим мальчиком, пока он не родился.
Они называли его овощем при рождении. Крестная мать Донни — медсестра, находилась в родильном зале.Она знала, что что-то не так. Первый врач, который его осмотрел, знал, что случилось. Он сказал: «Отведи его домой и люби его, и он умрет на твоих руках». Они сказали, что он будет слепым и слабослышащим, но это не так. И у него полноценная трисомия 18, он не мозаика.
Донни плохо функционирует. В этом году он трижды болел пневмонией. Он пережил инфекцию, вызванную C. difficile. У него лихорадка, проблемы с сердцем и кишечником, инфекции мочевыводящих путей, и он ломает свои хрупкие кости.Он не ходит и не разговаривает. Он весит 55 фунтов и больше не станет. Но он продолжает идти.
Он определенно не овощ. Он маленький человек. Он реагирует адекватно. Я использую отсасывающий аппарат, и когда я подношу часть к нему, он открывает рот. Он знает. Он узнает людей. Его лицо загорается, когда он видит знакомых. Я не совсем понимаю, что он такой редкий, но, по словам генетика, Донни считается самым старым человеком в мире с трисомией 18.
Он любит школу с особыми потребностями, и мне очень больно, потому что он стареет из-за всего. Он любит музыку и ходит в школьный автобус с другими детьми. Мне придется перестать называть его ребенком!
Он вокализирует. Я знаю, когда он намочил подгузник. Он издает радостные возгласы. Нам нравится смотреть игровые шоу по телевизору. Ему нравятся разноцветные огни, звук хлопков. Он улавливает волнение. Он лежит в большом кресле для ленивых мальчиков, а я сижу на диване рядом с ним.Нам нравится «Колесо фортуны», «Цена правильная», и давайте заключим сделку.
Я вижу свет в этих глазах. Донни меня поражает ».
Чтобы получить дополнительную информацию, связаться с другими или сделать пожертвование, см .:
Организация поддержки трисомии (СОФТ)
Фонд трисомии 18
Что такое синдром Эдварда?
Синдром Эдварда — это генетический дефект, который приводит к нескольким аномалиям в организме детей, рожденных с этим заболеванием.Младенцы с этим хромосомным заболеванием умирают вскоре после рождения. От этого состояния нет лекарства.
Синдром Эдварда, также известный как трисомия 18, по оценкам, поражает одного из пяти тысяч живорожденных. Вероятность того, что женщина родит ребенка с синдромом Эдварда, увеличивается с возрастом беременной.
Изображение предоставлено: Катерина Кон / Shutterstock.com
Хотя это заболевание вызывает дефект хромосом, это не наследственное заболевание.При трисомии 18 у ребенка есть три копии хромосомы номер 18 вместо необходимых двух.
Ошибка, вызывающая трисомию 18, обычно возникает во время производства спермы или яйцеклетки. В 90% случаев причиной дефекта является яйцо. Ошибка возникает при делении клеток и известна как мейотическая дизъюнкция.
Типы трисомии 18
Существует несколько различных типов трисомии 18, включая синдром Эдварда, мозаичную трисомию 18 и частичную трисомию 18.
Синдром Эдварда
Синдром Эдварда — самая тяжелая форма трисомии 18, так как пораженный ребенок, скорее всего, умрет вскоре после рождения.
У ребенка обычно есть множественные дефекты и аномалии органов, такие как маленькая деформированная голова, челюсть меньше среднего, сжатые кулаки с перекрытием пальцев, низко посаженные уши и экзомфалос, когда кишечник находится в мешочке. вне желудка. Младенцы, рожденные с синдромом Эдварда, также могут иметь проблемы с развитием сердца, легких и позвоночника.
Кредит изображения: Чали Чулапорнсири / Shutterstock
Мозаичная трисомия 18
При мозаичной трисомии 18 только некоторые клетки ребенка будут иметь дополнительную копию хромосомы 18. В зависимости от количества и типа пораженных клеток инвалидность будет отличаться от ребенка к ребенку.
Примерно семь из десяти детей с этим заболеванием могут прожить год и более. В редких случаях, когда инвалидность не очень выражена, человек может дожить до раннего взросления, но никогда не сможет действовать самостоятельно.В совокупности мозаичная трисомия 18 считается несколько менее тяжелой формой заболевания.
Частичная трисомия 18Частичная трисомия 18 считается наименее тяжелой формой заболевания. При частичной трисомии 18 присутствует только часть дополнительной хромосомы 18, а не вся дополнительная хромосома.
Тяжесть инвалидности ребенка при частичной трисомии 18 будет зависеть от части хромосомы, присутствующей в клетках, и количества клеток с хромосомным дефектом.Частичная трисомия 18 может быть унаследована, когда родитель несет перестройку генетического материала между двумя хромосомами, что называется сбалансированной транслокацией.
Диагностика трисомии 18
Скрининг на трисомию 18 проводится на сроке от 10 до 14 недель беременности. Комбинированный тест также проверяет синдром Дауна и синдром Патау.
Тем, кто окажется в группе высокого риска после комбинированного теста, будет рекомендовано пройти диагностический тест, чтобы подтвердить наличие синдрома Эдварда.Это делается путем проверки клеток ребенка на наличие дополнительной копии хромосомы 18.
Два способа получения образцов клеток включают взятие образца ворсин хориона и амниоцентез . В ворсинках хориона образцы клеток берутся из плаценты — ткани, соединяющей плод с матерью. При амниоцентезе берется образец околоплодных вод в утробе матери вокруг ребенка. Эта жидкость содержит клетки, которые потерял плод, и изучается для постановки точного диагноза.
Оба этих теста инвазивны и имеют небольшой риск выкидыша. В новом разработанном тесте используется только образец крови матери. Это неинвазивный метод пренатального тестирования, хотя он и дороже в применении.
Сканирование в середине беременности на сроке от 18 до 21 недели также предоставит информацию о наличии каких-либо физических отклонений у плода. Они называются врожденными аномалиями и часто сигнализируют о наличии серьезного заболевания.
Тотальная трисомия 18 (общая триссомия 18) Синдром Эдвардса Играть
Решение постдиагностики
Если у ребенка диагностирована трисомия 18, врач предложит прервать беременность, поскольку это серьезное заболевание, от которого нет лечения.